Ion channels are proteins that allow the passage of ions across biological membranes; particularly important cations include Na+, K+, and Ca++. Nerve impulse transmission involves Na+ and K+ channels; the advantages of quantum calculations are discussed.
- ion channels
- quantum calculations
- tunneling
- charge transfer
1. Introduction
Ion channels are proteins that allow the passage of ions, especially Na+, K+, and Ca++ across membranes. In this discussion, we will give most examples from a particular potassium channel. However, we will be primarily concerned with the question of the best means of obtaining information from calculations, which we will apply much more generally. In essence, there are two approaches to calculations on proteins, and channels in particular. The calculations can be done using classical or quantum physics, and we will argue that quantum physics is necessary. For one thing, there are certain quantum phenomena that are essentially impossible to simulate using classical physics. Of these, tunneling is almost certainly relevant to ion channels. Furthermore, certain energy terms have no classical analogues. While most of the energy has classical analogues (kinetic energy and Coulomb terms in the potential energy, for example), exchange and correlation energies are strictly quantum phenomena. Not all interactions scale with the same distance dependence; in short-range interactions (i.e., at roughly bonding distances, where quantum effects dominate), the dependence on interatomic distance does not scale in a simple manner. Coulomb interactions, on the other hand, scale as 1/r, and hence are much longer range. At interatomic distances greater than about 4 Å, Coulomb interactions dominate, and a classical approximation can be accurate; bonding interactions occur at somewhat shorter distances; those interactions that are inherently quantum are much more difficult to model classically, and switches in bonding, especially hydrogen bonding, are particularly difficult. For ion channels, therefore, where the interactions concern chemical bonding, and hydrogen bonding, it is necessary to use quantum calculations to be sure that all interactions are correctly modeled, with the correct distance dependence. We believe that proton tunneling, and proton transport, are critical. Proton transport is known to happen, but in gating models that have become standard, this is not considered relevant to channel function. Our calculations suggest that it is fundamental to channel function. The attempt to produce classical force fields that are sufficiently accurate to simulate channels, for example (other protein systems have similar requirements) has led to a great deal of effort [1][2][3]; this has succeeded only to a limited extent. This said, molecular dynamics (MD) calculations are much more commonly used (there is also a less commonly used classical physics alternative, Monte Carlo simulations, which do not do time dependence, but give a Boltzmann distribution of states). Simulations have been reviewed repeatedly (e.g., for ion channel interactions and other protein cases [4][5][6][7][8]).
There is also the possibility of doing QM/MM calculations (QM = quantum mechanics, MM = molecular mechanics, which is classical); this allows the key section of the protein to be done properly as a QM calculation, while the background is approximated much better than a vacuum; a pure QM calculation of a section of protein has a vacuum outside, albeit at a greater distance than the boundary between the QM and MM sections of the QM/MM calculations [9][10][11]. This is not the only multiscale method; the development of multiscale methods is ongoing, from many laboratories. The major advantage is that much more of the protein can be included, albeit at the MM level, while the main section of the calculation is done accurately at the QM level; the entire calculation is much faster than doing a large section of protein entirely at the QM level. The principal disadvantage is that one must create a boundary between the QM and the MM sections. Sometimes this can be done without causing major disruption. However, covalent bonds must usually be broken to create the boundary. This said, the smaller QM section makes the boundary closer to the key section. If the boundary introduces an extra dipole, or otherwise affects the local field at the main QM section, it can be worse than a vacuum with a somewhat more distant boundary. However, QM/MM is often considered as a useful compromise between the requirements for computer resources and the requirements for QM accuracy in calculation. For QM calculations that are intended to trace a path, whether of a K+ or H+, only differences are relevant, so that the boundary effects are largely subtracted out. This said, it would be better to include more, and it is not possible with limited computer resources.
In addition to splitting the calculation into QM and MM sections, other multiscale simulations are possible, in which different levels of computation can be applied to different parts of the system. For example, an outer part can be coarse-grained, in which groups of atoms are combined into a single item that can be treated with the same computer resources as a single atom. There are other possibilities, and a number of these have been explored [12][13][14][15].
This is a very limited summary of the reported calculations on ion channels—only a tiny fraction of the literature can be cited here. However, this sample suggests what alternate possibilities have been applied to ion channels and gives a suggestion of what alternatives exist for calculations on ion channels. We chose the full quantum mechanical approach, and we will outline our reasons below. However, as exascale computing becomes available, less approximation will be necessary, so that quantum calculations will become more practical. Although exascale computing is not yet generally available, it makes quantum calculations become of greater significance as the method of choice.
2. Our Calculations
It is worth considering the present state of affairs in our own calculations. We are using one node with 36 2.1 Gigaflop processors to get the optimized structure of systems with approximately 1000 atoms (maximum 1200 atoms). Typically, optimization at the (HF) level with an adequate basis set (we usually use 6–31G* or 6–31G**) takes about two months, allowing for some intervals and restarts. Generally, this gives a good enough structure for single point calculations at a higher level (typically density functional theory (DFT), using the exchange-correlational functional B3LYP, with the basis set 6–311G**). This gives the properties, especially the energy, at 0 K. The estimated accuracy is almost certainly within about 2 kBT (meaning T approximately 300 K, not 0K) for differences, which are what is needed for examining any mechanism in a channel. This is close enough to make comparisons meaningful, but not quite the best that could be done with a more accurate method. More to the point, the larger error is only taking about 1000 atoms. HF calculations scale as N4, where N is the number of atoms (actually the number of basis functions, but that scales linearly with number of atoms). Thus, the time required to do 2000 atoms, compared to 1000, is 16 times longer, outside the range of what is practical. Therefore, roughly 1000 atoms, is, at present, an approximate upper limit of the size of the system that can be optimized, without devoting more computer resources than can normally be allocated to one user.
Our calculations have other limitations. For one, the calculation is at 0 K. When comparing energy, this is not terrible, as the comparison is between two systems of the same size and approximately the same conformation. Therefore, the vibrations should be similar enough that the difference in energy at around 300 K would not normally be significantly different from the difference in ground state energy alone, which is what we calculate. There is another danger, that the system settles into a local minimum far from the global minimum. In one sense, we depend on this happening: we start the system with a proton or ion in a particular location, and do not expect that proton or ion to travel a considerable distance to find a different minimum. Instead, the optimization may, for example, move a proton to the other side of a hydrogen bond, and rotate a side chain as a consequence; if the calculation continued to find a global minimum every calculation would give the same result, after much too long a time to be practical. However, there exists a chance of convergence to a nearby very shallow, hence unimportant, minimum. Thus far, we have found no evidence of this, as the algorithm appears to move the system out of any minimum it comes close to, until it settles into convergence in a minimum too deep to leave. Nevertheless, it would be useful to confirm this, and we cannot at this point do so.
Gaussian09 [16], which is the computational suite we used the most, allows for the calculation of thermodynamic functions at finite temperature, in the harmonic approximation. However, the determination of the normal coordinates demands too much memory, using present resources, for systems above about 300 atoms. There is an additional question in doing such a calculation: normal coordinate analysis is a reasonable approximation for systems that are not too large. However, for a 1000 atom system, there should be many modes with anharmonic behavior. It would seem more likely that the appropriate approximation would be something more like a solid, with long wavelength modes in addition to more local, and more nearly harmonic modes of higher frequency. By analogy with solids, the latter would be more like Einstein modes. At this point, we do not have the means to examine this question, nor to determine whether it is significant or not. One thing that gives us confidence is that the modes should be very similar for two systems with the same structure, albeit with a proton shift or a water rotation. As a consequence, we continue to have confidence in comparing energy. This is supported by the fact that completely independent calculations differ by a small amount in reasonable directions; that is, each energy calculation starts from essentially the same position, with a proton shift or other small change; each optimization is independent. If the results were extremely sensitive to small perturbations, this would appear as large random changes in energy, something we do not see.
3. The Expected Effects of Computer Advances
With the coming of exascale computing, assuming that the computer facility must be shared as at present, and assuming that memory will be expanded proportionately, we should expect more than an order of magnitude increase in computing speed and capability. This also assumes that the software is upgraded to take advantage of the new power of the hardware. The use of graphics processors makes a direct comparison to present systems difficult. However, a speed up of perhaps 30-fold seems a reasonable estimate for an exascale system with GNU processors. This would allow 1000 atom jobs to be completed in about a day or two, as the time lost to restarts would be largely eliminated. We could then test enough cases, with enough overlap of the neighboring sections of the protein, that we could determine essentially every case we needed to. Second, we could do jobs of about 2500 atoms in a month, allowing for the N4 scaling of HF calculations; this would allow us to check that the 1000 atom jobs were correct with respect to boundaries. The boundaries would be sufficiently distant from the central region being computed that they should have little effect on the central region. If the results for the key regions were not significantly altered by moving the boundary of the computed region outward, then it would be possible to be sure the boundary was not affecting the result. If this were not confirmed, it would be possible to go back to month-long jobs, but with the boundaries distant enough that they should no longer have a major effect. We have reason to trust our present results, however, so we expect that this would not be necessary. We also note that taking the differences of related systems with the same number of atoms means that the energy differences, at least, are likely to cancel most of the boundary effects; they will be more accurate than the absolute values of individual computations.
There is another advantage to using QM: calculations at the DFT level, for example, using the natural bond orbital (NBO) method of Weinhold and coworkers [17], give the extent of charge transfer, a critical question for understanding the transfer of protons, and the conduction of ions through the pore of the channel. Knowing the charge also makes possible the calculation of the electric field locally, which must be important in all locations, except possibly where it is dominated by an externally applied field. Classical calculations must assume a charge distribution; with polarization, there can be a somewhat better approximation, but this is too time consuming to be used in large systems. However, even polarization does not allow for the correct treatment of charge transfer, which requires the consideration of the orbitals into which, or from which, electron density is transferred.
We observed above that calculations of thermodynamic functions are memory limited, at least in the normal modes’ approximation, but noted that this limitation may be lifted with exascale computers. With thermodynamic functions at finite temperature, we will be able to determine the equivalent of equilibrium constants for the population of states along the path of the protons, as well as for the ions in the selectivity filter. Knowing these, and estimating any remaining barriers, will allow the determination of rate constants in the standard manner.
Therefore, advances in computer facilities, at a level that is now beginning to be installed, should allow the straightforward use of quantum calculations for most purposes, without the approximations of using classical physics, or of installing boundaries between the QM and MM sections of the system. If we continue using the same QM methods as at present, we may still have the limitations of getting static results; these results may no longer be only at 0 K; size limitations of the present method are likely to be removed for most practical purposes, although it is likely that the harmonic approximation will not be so easily removed. Ideally, one would like to remove the limitation to static results as well by doing ab initio MD; however, it is not clear that even exascale computations would make this possible with systems large enough to be useful, for times of biological interest. If we have the states of the system with all reasonable proton positions, and the rate constants for transitions between pairs of conformations, we would have the equivalent. With the rate constants, we could find the populations of the sequential conformations as a function of time. We would also be able to determine alternate paths for the protons, which our present results strongly suggest exist, or a return (closing) path that differs from the path toward opening. We already have some evidence to support this. With MD, so far, it has not been possible to show that any path for a standard model can be reversed to return from open to closed, after a simulation of opening (or a reversible process in going back from closed to open). The channel must be able to return without error, perhaps thousands of times before the protein is replaced. If the channel operates at an average frequency of 10 Hz for 10 min, a likely minimum lifetime for the protein, this requires 6000 openings and closings. As far as we are aware, MD has yet to demonstrate that a simulated channel has ever returned to its starting conformation, except by using a directed simulation. While we have not explicitly considered returns, proton transfer uses a small number of degrees of freedom, and a return by at least one path, which we use for the forward path, is necessarily possible. There is another problem with MD, in that it generally uses a voltage or a field about an order of magnitude too large, in order to open the channel. If the correct field is used, the result is that the S4 segment fails to move, so that the standard model without proton transfer gives the same result as we obtain: nothing happens; or with the correct voltage, MD results, like ours, which do not have the S4 segment moving. The reason usually given for using the high voltage in MD is to allow the shorter times available for simulations to stand in for biological times. However, this introduces a new approximation: the effects of electric field are assumed to scale linearly with field. It is precisely those phenomena that are missed in a classical calculation that do not scale linearly, such as charge transfer, and proton switches in hydrogen bonds. For these reasons also, MD does not appear to be a useful method to characterize ion channels. This high a field exists near the second arginine, and that accounts for the entire voltage drop. 5 Å × 108 V m−1 = 50 mV, enough to depolarize the membrane. If the high field stretches across the entire membrane, one obtains a seriously unrealistic picture, but this does allow all the arginines to be dragged down, thereby creating the classical model, provided one accepts unrealistic conditions.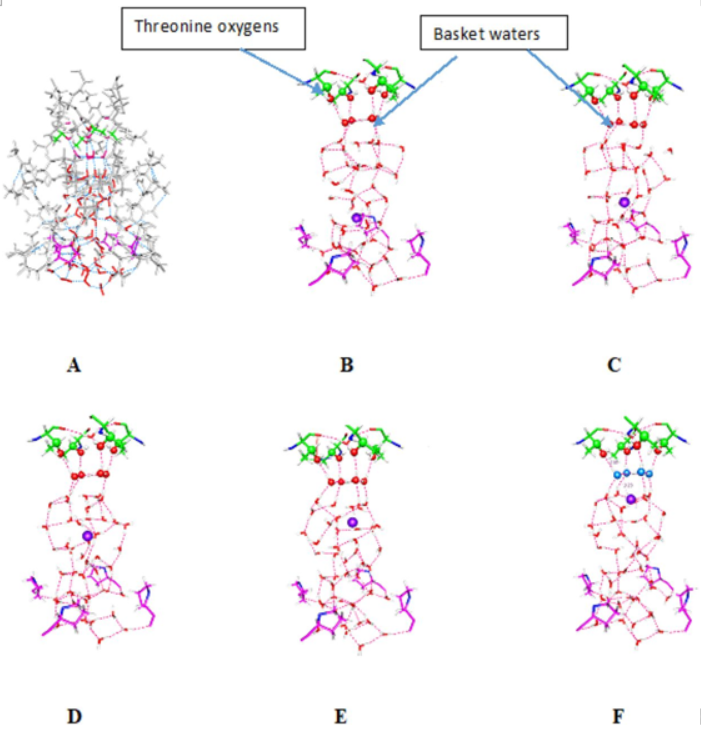 Figure. showing the cavity of a potassium ion channel, with one ion. In A, the protein is shown; in B through F, the protein is removed except for a small number of residues at the bottom marking the entrance to the cavity (the channel gate), and the exit to the selectivity filter at the top. The water and the ion can therefore be seen; the ion is shown as a purple sphere; in B it is at the lowest position in the pore, and in succeeding steps (C through F) the ion position moves up approximately 2 angstroms each step. The figure is reproduced from Kariev and Green bioRxiv (2018).
Figure. showing the cavity of a potassium ion channel, with one ion. In A, the protein is shown; in B through F, the protein is removed except for a small number of residues at the bottom marking the entrance to the cavity (the channel gate), and the exit to the selectivity filter at the top. The water and the ion can therefore be seen; the ion is shown as a purple sphere; in B it is at the lowest position in the pore, and in succeeding steps (C through F) the ion position moves up approximately 2 angstroms each step. The figure is reproduced from Kariev and Green bioRxiv (2018).
Overall, it seems necessary to do quantum calculations to obtain reasonably reliable results for ion channels. We can now do these calculations on systems of a size large enough to give useful insight into the properties of the channels. With the coming generation of computers, it seems likely that it will be practical to make the calculations sufficiently accurate, and on large enough systems, to confirm and extend these results to allow an essentially complete understanding of the properties and functioning of ion channels.
This entry is adapted from the peer-reviewed paper 10.3390/sym13040655
References
- Prajapati, J.D.; Mele, C.; Aksoyoglu, M.A.; Winterhalter, M.; Kleinekathöfer, U. Computational Modeling of Ion Transport in Bulk and through a Nanopore Using the Drude Polarizable Force Field. J. Chem. Inf. Model. 2020, 60, 3188–3203.
- Ocello, R.; Furini, S.; Lugli, F.; Recanatini, M.; Domene, C.; Masetti, M. Conduction and Gating Properties of the TRAAK Channel from Molecular Dynamics Simulations with Different Force Fields. J. Chem. Inf. Model. 2020, 60, 6532–6543.
- Furini, S.; Domene, C. Critical Assessment of Common Force Fields for Molecular Dynamics Simulations of Potassium Channels. J. Chem. Theory Comput. 2020, 16, 7148–7159.
- Sansom, M.S.P.; Bond, P.J.; Deol, S.S.; Grottesi, A.; Haider, S.; Sands, Z.A. Molecular simulations and lipid-protein interactions: Potassium channels and other membrane proteins. Biochem. Soc. Trans. 2005, 33, 916–920.
- Allison, J.R. Computational methods for exploring protein conformations. Biochem. Soc. Trans. 2020, 48, 1707–1724.
- Bumbacila, B.; Putz, M.V. Monte Carlo Molecular Simulations; Apple Academic Press Inc.: Palm Bay, FL, USA, 2020.
- Horsewill, A.J. Quantum Molecular Tunneling Studied by Field-cycling NMR. New Dev. NMR 2019, 18, 405–426.
- Sharma, V.K.; Srinivasan, H.; Sakai, V.G.; Mitra, S. Dioctadecyldimethylammonium bromide, a surfactant model for the cell membrane: Importance of microscopic dynamics. Struct. Dyn. 2020, 7, 051301.
- Bucher, D.; Guidoni, L.; Carloni, P.; Rothlisberger, U. Coordination Numbers of K+ and Na+ Ions Inside the Selectivity Filter of the KcsA Potassium Channel: Insights from First Principles Molecular Dynamics. Biophys. J. 2010, 98, L47–L49.
- Cholasseri, R.; De, S. Dual-Site Binding of Quaternary Ammonium Ions as Internal K+-Ion Channel Blockers: Nonclassical (C–H··· O) H Bonding vs Dispersive (C–H··· H–C) Interaction. J. Phys. Chem. B 2021, 125, 86–100.
- Carnevale, V.; Fiorin, G.; Levine B., G.; Degrado W., F.; Klein, M. Multiple Proton Confinement in the M2 Channel from the Influenza A Virus. J. Phys. Chem.C Nanomater. Interfaces 2010, 114, 20856–20863.
- Masetti, M.; Berti, C.; Ocello, R.; Di Martino, G.P.; Recanatini, M.; Fiegna, C.; Cavalli, A. Multiscale Simulations of a Two-Pore Potassium Channel. J. Chem. Theory Comput. 2016, 12, 5681–5687.
- Qi, Y.; Cheng, X.; Han, W.; Jo, S.; Schulten, K.; Im, W. CHARMM-GUI PACE CG Builder for Solution, Micelle, and Bilayer Coarse-Grained Simulations. J. Chem. Inf. Model. 2014, 54, 1003–1009.
- Vianello, R.; Domene, C.; Mavri, J. The Use of Multiscale Molecular Simulations in Understanding a Relationship between the Structure and Function of Biological Systems of the Brain: The Application to Monoamine Oxidase Enzymes. Front. Neurosci. 2016, 10, 327.
- Von Wegner, F.; Wieder, N.; Fink, R.H.A. Simulation strategies for calcium microdomains and calcium-regulated calcium channels. Adv. Exp. Med. Biol. 2012, 740, 553–567.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01. 2009; Gaussian, Inc.: Wallingford, CT, USA, 2013.
- Weinhold, F. Natural bond orbital analysis: A critical overview of relationships to alternative bonding perspectives. J. Comput. Chem. 2012, 33, 2363–2379.