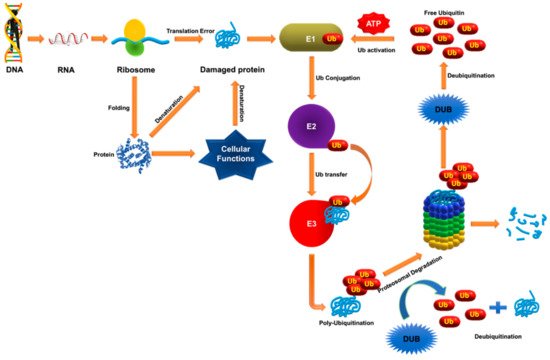
6. Deubiquitinases as Emerging Targets against Hematological Malignancies
Ubiquitination and deubiquitination play critical roles in various biological pathways closely associated with development of various cancers. Although knowledge about the precise role of DUBs in cancer pathology is limited, the list of DUBs that are altered genetically in human cancer cases has grown rapidly in recent years [
162]. Recent studies have presented DUBs as a genuine oncogene and tumor suppressor. DUBs that regulate cellular expression and turnover of oncogenic protein in various hematological malignancies have been identified. In recent years, inhibitors of the UPS have emerged as therapeutic targets for the treatment of various cancers. However, application of most of these inhibitors are hampered by low efficacy in hematological malignancies [
163]. The ability of DUBs to modulate the fate of a protein specifically and selectively gives them an advantage over targeting the UPS. For example, DUBs may increase or maintain levels of a tumor suppressor protein by decreasing its degradation by UPS or boost pathogenesis by reversing the fate of oncogenic proteins in the cell [
26]. Considering the advantages and ease of developing inhibitors over enzyme activators, research into the development of DUB inhibitors against hematological malignancies has been emphasized.
Recent approaches to targeting DUBs through various small-molecule inhibitors have produced promising results against various hematological malignancies. The novel regulatory particle b-AP15 together with lenalidomide, or dexamethasone, induces synergistic anti-MM activity [
140]. According to recent studies, b-AP15 and VLX1570 could also be a potential therapy for leukemia and WM by inhibiting 19S proteasome-associated DUBs such as USP14/UCHL5 and inducing tumor-cell apoptosis. VLX1570 along with dexamethasone was in clinical trial phase I/II but was terminated because of dose-limiting toxicity [
164]. Another potent DUB inhibitor WP1130, previously known as Degrasyn, targets deubiquitinases such as USP5, USP9X, USP14, USP24, and UCHL5. Recent studies concluded that WP1130-mediated inhibition of USP9X increases ubiquitination of an anti-apoptotic protein MCL1 which is highly expressed in drug-resistant MM tumors [
127,
165]. The rapid degradation of MCL1 results in an increase in the sensitivity of these tumor cells to chemotherapy [
26,
163,
166]. USP24, which is closely related to USP9X, also plays a critical role in the survival of myeloma B cells by regulating MCL1 protein levels. Peterson and colleagues suggested that dual inhibition of USP9X and USP24 by WP1130 provides greater anti-myeloma activity. However, they developed an inhibitor that is three times as effective called EOAI3402143 (G9), which also had an improved therapeutic index. G9 inhibits USP5, increasing p53 accumulation and therefore is a promising approach to treating B-cell malignancies [
127,
167]. At present, studies of USP7 and USP10 inhibitors (HBX19818 and P22077), which play key roles in many cellular processes, are under way. A lead-like inhibitor, HBX41108 and HBX19818, has been found to inhibit the catalytic activity of USP7 and induce p53-dependent apoptosis [
168,
169].
Preclinical data demonstrated the anti-tumor efficacy of P5091 with lenalidomide, histone deacetylase inhibitor SAHA, or dexamethasone by inducing tumor-cell apoptosis in MM disease models [
118]. It also stabilizes p53 by inhibiting USP7 mediated-deubiquitination of MDM2, which degrades p53 tumor suppressors [
115,
170,
171], thus inhibiting cancer cell proliferation. HBX19818, P5091, as well as their analogs, including P045204, P22077, and HBX41108 have been shown to be potent inhibitors of USP7 [
172]. P217564, a second-generation inhibitor, binds to the active site of USP7, inhibiting its activity [
173]. Along with USP7, P22077 and HBX19818 has also been reported to inhibit USP10, promoting degradation of FLT3-mutant AML cells [
174]. A small-molecule inhibitor, spautin-1 (for
specific and potent
autophagy
inhibitor-1), inhibits autophagy, and two DUBs, USP10 and USP13, which deubiquitinates two tumor suppressors, Beclin1, a subunit of Vps34 complexes, and p53 [
175].
Despite the development of several inhibitors against DUBs, none of them have gone on to clinical trials, likely due to the complex structure of the catalytic domain of DUBs that share similarities with other DUB family members. Upon ubiquitin-binding, the active sites of DUBs also undergo conformational changes, posing a challenge when designing as well as binding the inhibitor to its specific DUB. The UPS consists of two regulatory enzymes, E3 ligase, and DUBs, which work as a complex, and understanding the dynamics of the complex, not just the DUBs, is essential to engineering a specific inhibitor. Moreover, it has been well documented that diverse DUBs play crucial roles in many cellular processes. Future research should be channeled toward developing small-molecule inhibitors that target the conserved catalytic cysteine of DUBs with stable and selective substrate-binding efficacy using new technologies. A high-throughput screening method should be made available with which researchers can determine the combination of DUBs and/or inhibitors that best modulate active pathways in cancer.
A better understanding of regulatory DUBs involved in inhibition or activation of hematopoietic processes and pathologies is expected to open new frontiers in the development of novel therapeutic drugs that target hematological malignancies and disorders. The goals are to enhance our understanding of dysregulated DUBs in hematopoiesis; design new therapeutic targets, and establish biomarkers that could be used in diagnosis and prognosis.
7. Conclusions
Since its discovery, the UPS has emerged as a key regulator of various proteins and factors involved in hematopoiesis, erythropoiesis, and angiogenesis. The roles of E1, E2, and E3 enzymes in governing the various pathways involved in hematopoietic regulation and pathologies have been studied extensively, but knowledge about the reversal of the activity of DUBs and their involvement in various hematological processes is limited. Several studies provide insight into dysregulated functioning of DUBs in various hematopoietic cells, which contribute to hematological pathologies. In this review, we described various DUBs that directly or indirectly regulate various hematopoietic processes.
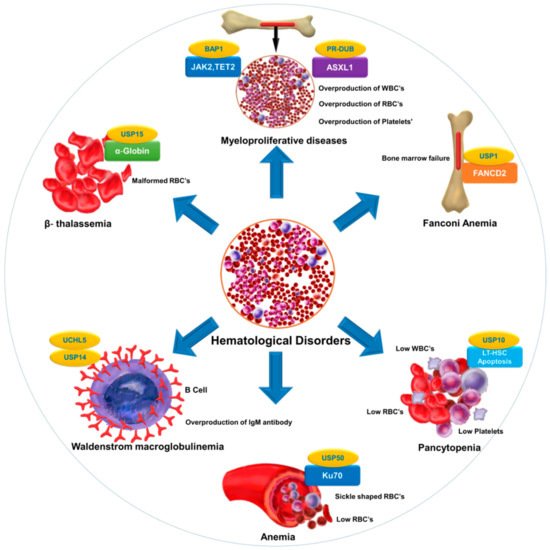
Selective inhibition or overexpression of DUBs has helped elucidate their roles in hematopoiesis, erythropoiesis, angiogenesis, and related abnormalities. Potent selective inhibitors of DUBs have shown promise for the treatment of hematological malignancies. Hematological disorders are the result of one or more malfunctioning components in the blood caused by intrinsic factors. DUBs are thought to play an important role in the etiology of various diseases and disorders and are therefore attractive drug targets. However, limited knowledge about substrate specificity and the molecular mechanisms of action of DUBs currently restricts their utility as novel therapeutic targets ().
Figure 2. DUB as a novel target in hematological diseases. DUBs regulate the level and function of a protein by catalyzing removal of ubiquitin from the substrate protein. Dysregulation of DUBs contributes to the pathogenesis of various hematological disorders. The figure illustrates different hematological disorders and the associated DUBs that can provide novel targets for therapeutic interventions to treat these disorders.
provides a list of DUBs associated with blood disorders. Identification of other DUBs involved in the pathogenesis of other hematological disorders and more complete insight into the regulatory mechanisms of DUBs that govern disease progression will provide new perspectives in therapeutics.
Table 1. List of DUBs involved in hematological disorders.
| Disorder |
Associated Substrate |
Regulatory DUB |
Reference |
| Fanconi anemia |
FANCD2 |
USP1 |
[63,176] |
| Anemia |
Ku70 |
USP50 |
[8] |
| β-thalassemia |
α-globin |
USP15 |
[177] |
| Pancytopenia |
Reduction in LT-HSC |
USP10 |
[31] |
| Myeloproliferative diseases |
ASXL1, EZH2,JAK2,TET2 |
PR-DUB,BAP1 |
[178,179] |
| Waldenstrom macroglobulinemia (WM) |
Overexpression of USP14 and UCHL5 in drug-resistant WM-tumor cells |
USP14 and UCHL5 |
[137,180,181] |
| Bone marrow failure |
B-cell factor 1 (Ebf1), paired box 5 (Pax5), and other B-lymphoid genes |
MYSM1 |
[45,182] |
| Malaria |
CD8+ T cells |
CYLD |
[162,183] |
Table 1. List of DUBs involved in hematological disorders.
Although DUBs may play important roles in the regulation of hematopoiesis, many questions about their involvement remain unexplored. The exact DUBs-mediated signaling pathways responsible for the progression of hematological disorders have yet to be elucidated. Much of this knowledge gap stems from the ability of a given DUB to regulate several hundreds of protein substrates. At the same time, a given protein substrate can be regulated by more than one DUB.
Despite progress toward the development of DUB inhibitors, a lack of specificity limits clinical application of a wide range of DUBs [
112]. This limitation may be overcome by improved understanding of self- versus trans-regulation of DUBs and applying this information to inhibitor synthesis prior to applying such DUB inhibitors to preclinical and clinical research. Furthermore, another possible mechanism of “dubbing” DUBs in cases where a particular DUB itself is regulated by another DUB should also be seriously considered [
13]. Expanding research on dubbing DUBs is expected to offer novel insights into understanding DUB regulatory networks and implementing innovative strategies to uncover and develop molecular therapies to treat hematopoietic diseases.
The DUBs discussed in this review raise the question of whether there is a master DUB regulator that can deubiquitinate multiple DUBs. However, conventional DUB-mediated therapeutic approaches involve targeting DUBs that contribute to hematological pathologies. Dubbing DUBs may allow for screening for a master DUB that regulates the key DUBs implicated in hematological pathologies. Once such master DUBs are identified, the task of generating selective and specific inhibitors of DUBs implicated in hematologic disorders may become easier. We hypothesize that mapping an exclusive inter-DUB regulatory network combined with wide proteomic-scale screening of crucial DUBs will increase our understanding of several unknown links that may be related to the DUB regulatory network in hematological malignancies.
The role of the DUBs in HSC maintenance and differentiation remains an active area of research. Further investigation into the localization and substrate specificity of DUBs, their interactions with other hematopoietic factors, and other data gaps will improve our knowledge about their role in hematopoiesis. Research in this direction will facilitate the development of specific inhibitors against the key DUBs implicated in hematopoiesis. Doing so, while minimizing undesirable side effects, should lead to exciting new opportunities in treating hematologic malignancies.