Inflammation is the main driver of tumor initiation and progression in colitis-associated colorectal cancer (CAC). Recent findings have indicated that the signal transducer and activator of transcription 6 (STAT6) plays a fundamental role in the early stages of CAC, and STAT6 knockout (STAT6−/−) mice are highly resistant to CAC development. Regulatory T (Treg) cells play a major role in coordinating immunomodulation in cancer; however, the role of STAT6 in the induction and function of Treg cells is poorly understood. To clarify the contribution of STAT6 to CAC, STAT6−/− and wild type (WT) mice were subjected to an AOM/DSS regimen, and the frequency of peripheral and local Treg cells was determined during the progression of CAC. When STAT6 was lacking, a remarkable reduction in tumor growth was observed, which was associated with decreased inflammation and an increased number of CD4+CD25+Foxp3+ cells. STAT6 has a direct role in the induction and function of Treg cells during CAC development.
- STAT6
- colorectal cancer
- regulatory T cells
- colitis-associated-cancer
1. Introduction
2. The Absence of STAT6 Increases the Number of CD4+CD25+Foxp3+ Cells in Circulation and Spleen during the Early Stages of CAC Development
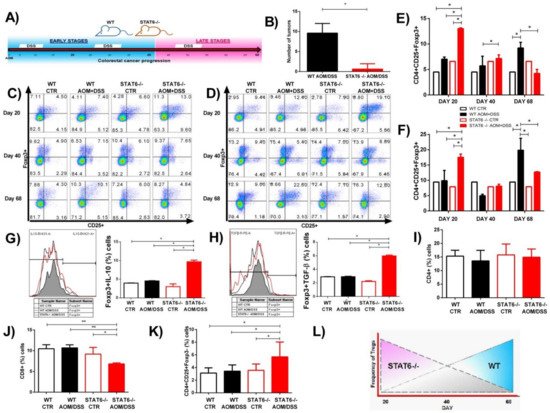
3. STAT6 Deficiency Increase the Accumulation of Treg Cells in the Colon in Early CAC
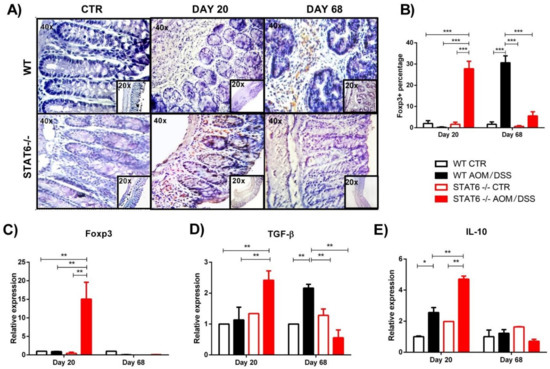
4. Discussion
This entry is adapted from the peer-reviewed paper 10.3390/ijms22084049
References
- Siegel, R.; DeSantis, C.; Jemal, A. Colorectal cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 104–117.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 68, 394–424.
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081.
- Van Der Kraak, L.; Gros, P.; Beauchemin, N. Colitis-associated colon cancer: Is it in your genes? World J. Gastroenterol. 2015, 21, 11688–11699.
- Hebenstreit, D.; Wirnsberger, G.; Horejs-Hoeck, J.; Duschl, A. Signaling mechanisms, interaction partners, and target genes of STAT6. Cytokine Growth Factor Rev. 2006, 17, 173–188.
- Delgado-Ramirez, Y.; Colly, V.; Gonzalez, G.V.; Leon-Cabrera, S. Signal transducer and activator of transcription 6 as a target in colon cancer therapy (Review). Oncol. Lett. 2020, 20, 455–464.
- Andre, F.; Arnedos, M.; Baras, A.S.; Baselga, J.; Bedard, P.L.; Berger, M.F.; Bierkens, M.; Calvo, F.; Cerami, E.; Chakravarty, D.; et al. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831.
- Uhlén, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507.
- Wang, C.-G.; Ye, Y.-J.; Yuan, J.; Liu, F.-F.; Zhang, H.; Wang, S. EZH2 and STAT6 expression profiles are correlated with colorectal cancer stage and prognosis. World J. Gastroenterol. 2010, 16, 2421–2427.
- Wick, E.C.; Leblanc, R.E.; Ortega, G.; Robinson, C.; Platz, E.; Pardoll, E.M.; Iacobuzio-Donahue, C.; Sears, C.L. Shift from pStat6 to pStat3 predominance is associated with inflammatory bowel disease-associated dysplasia. Inflamm. Bowel Dis. 2012, 18, 1267–1274.
- Li, B.H.; Yang, X.Z.; Li, P.D.; Yuan, Q.; Liu, X.H.; Yuan, J.; Zhang, W.J. IL-4/Stat6 activities correlate with apoptosis and metastasis in colon cancer cells. Biochem. Biophys. Res. Commun. 2008, 369, 554–560.
- Lin, Y.; Li, B.; Yang, X.; Liu, T.; Shi, T.; Deng, B.; Zhang, Y.; Jia, L.; Jiang, Z.; He, R. Non-hematopoietic STAT6 induces epithelial tight junction dysfunction and promotes intestinal inflammation and tumorigenesis. Mucosal Immunol. 2019, 12, 1304–1315.
- Leon-Cabrera, S.A.; Molina-Guzman, E.; Delgado-Ramirez, Y.G.; Vázquez-Sandoval, A.; Ledesma-Soto, Y.; Pérez-Plasencia, C.G.; Chirino, Y.I.; Delgado-Buenrostro, N.L.; Rodríguez-Sosa, M.; Vaca-Paniagua, F.; et al. Lack of STAT6 Attenuates Inflammation and Drives Protection against Early Steps of Colitis-Associated Colon Cancer. Cancer Immunol. Res. 2017, 5, 385–396.
- Olguín, J.E.; Medina-Andrade, I.; Rodríguez, T.; Rodríguez-Sosa, M.; Terrazas, L.I. Relevance of Regulatory T Cells during Colorectal Cancer Development. Cancers 2020, 12, 1888.
- Liu, Z.; Huang, Q.; Liu, G.; Dang, L.; Chu, D.; Tao, K.; Wang, W. Presence of FOXP3+Treg cells is correlated with colorectal cancer progression. Int. J. Clin. Exp. Med. 2014, 7, 1781–1785.
- Argon, A.; Vardar, E.; Kebat, T.; Ömer, E.; Erkan, N. The Prognostic Significance of FoxP3+ T Cells and CD8+ T Cells in Colorectal Carcinomas. J. Environ. Pathol. Toxicol. Oncol. 2016, 35, 121–131.
- Reimers, M.S.; Engels, C.C.; Putter, H.; Morreau, H.; Liefers, G.J.; Van De Velde, C.J.H.; Kuppen, P.J.K. Prognostic value of HLA class I, HLA-E, HLA-G and Tregs in rectal cancer: A retrospective cohort study. BMC Cancer 2014, 14, 486.
- Soh, J.S.; Jo, S.I.; Lee, H.; Do, E.-J.; Hwang, S.W.; Park, S.H.; Ye, B.D.; Byeon, J.-S.; Yang, S.-K.; Kim, J.H.; et al. Immunoprofiling of Colitis-associated and Sporadic Colorectal Cancer and its Clinical Significance. Sci. Rep. 2019, 9, 1–10.
- Vlad, C.; Kubelac, P.; Fetica, B.; Vlad, D.; Irimie, A.; Achimas-Cadariu, P. The prognostic value of FOXP3+ T regulatory cells in colorectal cancer. J. BUON 2015, 20, 114–119.
- Saito, T.; Nishikawa, H.; Wada, H.; Nagano, Y.; Sugiyama, D.; Atarashi, K.; Maeda, Y.; Hamaguchi, M.; Ohkura, N.; Sato, E.; et al. Two FOXP3+CD4+ T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 2016, 22, 679–684.
- Olguín, J.E.; Medina-Andrade, I.; Molina, E.; Vázquez, A.; Pacheco-Fernández, T.; Saavedra, R.; Pérez-Plasencia, C.; Chirino, Y.I.; Vaca-Paniagua, F.; Arias-Romero, L.E.; et al. Early and Partial Reduction in CD4+Foxp3+ Regulatory T Cells during Colitis-Associated Colon Cancer Induces CD4+ and CD8+ T Cell Activation Inhibiting Tumorigenesis. J. Cancer 2018, 9, 239–249.
- Bruns, H.A.; Schindler, U.; Kaplan, M.H. Expression of a Constitutively Active Stat6 In Vivo Alters Lymphocyte Homeostasis with Distinct Effects in T and B Cells. J. Immunol. 2003, 170, 3478–3487.
- Sanchez-Guajardo, V.; Tanchot, C.; O’Malley, J.T.; Kaplan, M.H.; Garcia, S.; Freitas, A.A. Agonist-driven development of CD4+CD25+Foxp3+ regulatory T cells requires a second signal mediated by Stat6. J. Immunol. 2007, 178, 7550–7556.
- Zorn, E.; Nelson, E.A.; Mohseni, M.; Porcheray, F.; Kim, H.; Litsa, D.; Bellucci, R.; Raderschall, E.; Canning, C.; Soiffer, R.J.; et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 2006, 108, 1571–1579.
- Takaki, H.; Ichiyama, K.; Koga, K.; Chinen, T.; Takaesu, G.; Sugiyama, Y.; Kato, S.; Yoshimura, A.; Kobayashi, T. STAT6 inhibits TGF-beta 1-mediated Foxp3 induction through direct binding to the Foxp3 promoter, which is reverted by retinoic acid receptor. J. Biol. Chem. 2008, 283, 14955–14962.
- Dorsey, N.J.; Chapoval, S.P.; Smith, E.P.; Skupsky, J.; Scott, D.W.; Keegan, A.D. STAT6 controls the number of regulatory T cells in vivo, thereby regulating allergic lung inflammation. J. Immunol. 2013, 191, 1517–1528.
- Rosen, M.J.; Chaturvedi, R.; Washington, M.K.; Kuhnhein, L.A.; Moore, P.D.; Coggeshall, S.S.; McDonough, E.M.; Weitkamp, J.-H.; Singh, A.B.; Coburn, L.A.; et al. STAT6 deficiency ameliorates severity of oxazolone colitis by decreasing expression of claudin-2 and Th2-inducing cytokines. J. Immunol. 2013, 190, 1849–1858.
- Jayakumar, A.; Bothwell, A.L. Stat6 Promotes Intestinal Tumorigenesis in a Mouse Model of Adenomatous Polyposis by Expansion of MDSCs and Inhibition of Cytotoxic CD8 Response. Neoplasia 2017, 19, 595–605.
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532.
- Ling, Z.-A.; Zhang, L.-J.; Ye, Z.-H.; Dang, Y.-W.; Chen, G.; Li, R.-L.; Zeng, J.-J. Immunohistochemical distribution of FOXP3+ regulatory T cells in colorectal cancer patients. Int. J. Clin. Exp. Patho. 2018, 11, 1841–1854.
- Ma, Q.; Liu, J.; Wu, G.; Teng, M.; Wang, S.; Cui, M.; Li, Y. Co-expression of LAG3 and TIM3 identifies a potent Treg population that suppresses macrophage functions in colorectal cancer patients. Clin. Exp. Pharmacol. Physiol. 2018, 45, 1002–1009.
- Norton, S.E.; Ward-Hartstonge, K.A.; McCall, J.L.; Leman, J.K.H.; Taylor, E.S.; Munro, F.; Black, M.A.; Groth, B.F.D.S.; McGuire, H.M.; Kemp, R.A. High-Dimensional Mass Cytometric Analysis Reveals an Increase in Effector Regulatory T Cells as a Distinguishing Feature of Colorectal Tumors. J. Immunol. 2019, 202, 1871–1884.
- Clarke, S.L.; Betts, G.J.; Plant, A.; Wright, K.L.; El-Shanawany, T.M.; Harrop, R.; Torkington, J.; Rees, B.I.; Williams, G.T.; Gallimore, A.M.; et al. CD4+CD25+FOXP3+ Regulatory T Cells Suppress Anti-Tumor Immune Responses in Patients with Colorectal Cancer. PLoS ONE 2006, 1, e129.
- Hanke, T.; Melling, N.; Simon, R.; Sauter, G.; Bokemeyer, C.; Lebok, P.; Terracciano, L.M.; Izbicki, J.R.; Marx, A.H. High intratumoral FOXP3+ T regulatory cell (Tregs) density is an independent good prognosticator in nodal negative colorectal cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 8227–8235.
- Shang, B.; Liu, Y.; Jiang, S.J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3(+) regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 1–9.
- Saito, T.; Yamashita, K.; Tanaka, K.; Yamamoto, K.; Makino, T.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; Wada, H.; Nishikawa, H.; et al. Impact of tumor infiltrating effector regulatory T cells on the prognosis of colorectal cancers. Cancer Sci. 2021, 112, 390.
- Fantini, M.C.; Favale, A.; Onali, S.; Facciotti, F. Tumor Infiltrating Regulatory T Cells in Sporadic and Colitis-Associated Colorectal Cancer: The Red Little Riding Hood and the Wolf. Int. J. Mol. Sci. 2020, 21, 6744.
- Omenetti, S.; Pizarro, T.T. The Treg/Th17 Axis: A Dynamic Balance Regulated by the Gut Microbiome. Front. Immunol. 2015, 6, 639.
- Liu, Y.-J.; Tang, B.; Wang, F.-C.; Tang, L.; Lei, Y.-Y.; Luo, Y.; Huang, S.-J.; Yang, M.; Wu, L.-Y.; Wang, W.; et al. Parthenolide ameliorates colon inflammation through regulating Treg/Th17 balance in a gut microbiota-dependent manner. Theranostics 2020, 10, 5225–5241.
- Ladoire, S.; Martin, F.; Ghiringhelli, F. Prognostic role of FOXP3+ regulatory T cells infiltrating human carcinomas: The paradox of colorectal cancer. Cancer Immunol. Immunother. 2011, 60, 909–918.
- Maloy, K.J.; Salaun, L.; Cahill, R.; Dougan, G.; Saunders, N.J.; Powrie, F. CD4(+)CD25(+) T-R cells suppress innate immune pathology through cytokine-dependent mechanisms. J. Exp. Med. 2003, 197, 111–119.
- Mottet, C.; Uhlig, H.H.; Powrie, F. Cutting Edge: Cure of Colitis by CD4+CD25+ Regulatory T Cells. J. Immunol. 2003, 170, 3939–3943.
- Watanabe, K.; Rao, V.P.; Poutahidis, T.; Rickman, B.H.; Ohtani, M.; Xu, S.; Rogers, A.B.; Ge, Z.; Horwitz, B.H.; Fujioka, T.; et al. Cytotoxic-T-Lymphocyte-Associated Antigen 4 Blockade Abrogates Protection by Regulatory T Cells in a Mouse Model of Microbially Induced Innate Immune-Driven Colitis. Infect. Immun. 2008, 76, 5834–5842.
- Canavan, J.B.; Scotta, C.; Vossenkamper, A.; Goldberg, R.; Elder, M.J.; Shoval, I.; Marks, E.; Stolarczyk, E.; Lo, J.W.; Powell, N.; et al. Developing in vitro expanded CD45RA(+) regulatory T cells as an adoptive cell therapy for Crohn’s disease. Gut 2016, 65, 584–594.
- Clough, J.N.; Omer, O.S.; Tasker, S.; Lord, G.M.; Irving, P.M. Regulatory T-cell therapy in Crohn’s disease: Challenges and advances. Gut 2020, 69, 942–952.
- Desalegn, G.; Pabst, O. Inflammation triggers immediate rather than progressive changes in monocyte differentiation in the small intestine. Nat. Commun. 2019, 10, 1–14.
- Chu, K.-H.; Lin, S.-Y.; Chiang, B.-L. STAT6 Pathway Is Critical for the Induction and Function of Regulatory T Cells Induced by Mucosal B Cells. Front. Immunol. 2021, 11, 11.