Hereditary angioedema is a rare inherited disorder characterized by recurrent episodes of the accumulation of fluids outside of the blood vessels, causing rapid swelling of tissues in the hands, feet, limbs, face, intestinal tract, or airway.
- HAE
- C1-INH-HAE
- nC1-INH-HAE
- SERPING1
- F12
- PLG
1. Introduction
Angioedema is characterized by a localized, self-limiting, and transient subcutaneous or submucosal swelling, which can present with or without episodes of urticaria, and usually subsides within 24–37 h. The clinical expression is highly variable, from asymptomatic individuals to patients suffering from disabling and life-threatening attacks with a demonstrated humanistic and economic burden [1]. Manifestations may imply swelling of the extremities and superficial regions of the face; affect the gastrointestinal tract, because of edema of the bowel wall, and cause severe cramping abdominal pain, nausea, vomiting and, sometimes, diarrhea; and occasionally compromise breathing, including laryngeal edema and/or severe tongue/pharyngeal edema so that secretions cannot be handled [2]. The severity and frequency of acute attacks of angioedema are variable, ranging from once/year to three attacks/week [3]. Although a series of predisposing circumstances, including trauma, fluctuating hormone level (particularly increased estrogen), infection, and severe emotional stress have been identified, most acute episodes of angioedema seem apparently spontaneous. In the general population, an acute episode of angioedema has been estimated to occur in up to 7.4% of subjects during their lifetime [4]. Thus, after excluding cases of angioedema with identifiable cause or wheals, one person in a hundred is likely to have an episode of angioedema. Approximately 0.05% of them have been estimated to suffer from recurrent/nonallergic angioedema [5].
2. Hereditary Angioedema
Hereditary angioedema (HAE) is a genetic disorder that predisposes an individual to develop vasogenic edema. Prevalence of HAE has been reported to be 1 in 10,000 to 1 in 150,000 [6]. HAE shows no ethnic- or sex-based differences but tends to be more severe in women [2,7]. The pathogenesis of HAE involves the accumulation of extravascular fluid in various tissues via a non-inflammatory and non-allergic mechanism. Clinical manifestations include abrupt onset swelling around the eyes, face and extremities; pain in theabdomen (as a result of bowel edema), and laryngeal edema leading to hoarseness of voice, breathing difficulty, and occasionally death [8,9].
Several disorders may manifest with subcutaneous or submucosal swelling. The presence of severe swelling can be mistaken for an allergic reaction or acute abdominal condition. Misdiagnosis can lead to ineffective therapies and unnecessary surgeries [10]. In 1963, Donaldson and Evans discovered that HAE was caused by a genetic deficiency of the C1 inhibitor (C1-INH) [11]. Since then, many large studies have established that the most common type of hereditary angioedema (HAE) is the result of gene mutations resulting in reduced C1-INH functional plasma. This results in two HAE variants of C1-INH-HAE: type 1 and type 2. C1-INH type 1 results from a failure to synthesize C1-INH, whereas in type 2, an abnormal, non-functional protein is synthesized.
The increase in vascular permeability that causes angioedema in HAE is related to the mediators of the contact system or kallikrein–kinin pathway. C1-INH regulates the contact system through the inhibition of plasma kallikrein and coagulation factor FXIIa. The loss of the inhibitory activity of C1-INH leads to bradykinin overproduction. It has been documented that an increased release of bradykinin is the cause of angioedema via its action on B2 receptors leading to an increase in vascular permeability [12,13]. Moreover, a dominantly inherited disease has been described that has a similar clinical picture to C1-INH-HAE, but with normal C1-INH level and activity. This new type of HAE has no mutation in the SERPING1 gene and it is classified as nC1-INH-HAE (Figure 1).
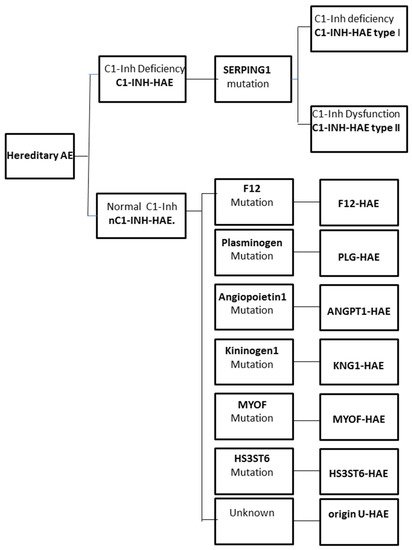
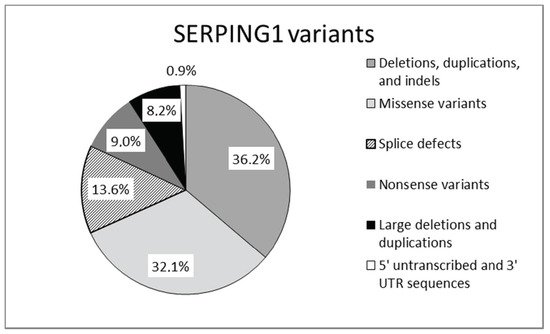
2.1. Genetics of C1-INH-HAE
2.2. The Genetics of nC1-INH-HAE
| F12 | T328K T328R c.971_1018 + 24del72 c.892_909dup |
| PLG | K330E |
| ANGPT1 | A119S A8V Q370H |
| KNG1 | M379K |
| MYOF | R217S |
| HS3ST6 | T144S |
2.3. Disease-Modifying Factors
The high variability in clinical expression between patients with the same mutation led to the hypothesis that mutations in these and other related genes could be potential modifiers in the clinical phenotype of patients with known genetic causes of HAE. Several polymorphisms in different genes and their effects on the clinical phenotype of patients were analyzed. These included p.Y244C, p.G354R, and p.T916M in the ACE gene; p.C548Y in the KLKB1 (kallikrein) gene; and p.D287N in the NOS3 (nitric oxide synthase) gene [43]. However, the role of these polymorphisms on modification in the clinical phenotype of HAE remains to be clarified. Other disease-modifying factors, F12 or KLKB1 gene polymorphisms, have been studied to explain clinical variability of C1-INH-HAE or nC1-INH-HAE. The possible associations of the functional promoter F12-46C/T polymorphism (rs1801020) with clinical features of C1-INH-HAE and the SERPING1 mutational status have been investigated. The F12-46C/T carriage acts as an independent modifier of C1-INH-HAE severity regardless of SERPING1 mutational status [44,45]. The c.-4C/T polymorphism (rs1801020) in the 5-UTR region of the F12 gene was shown to significantly influence the degree of contact system activation and the clinical severity of the disease. Patients carrying FXII-HAE (p.Thr309Lys variant) and c.-46CC genotype exhibited more severe and frequent manifestations of the disease [46].
The c.428G/A (rs3733402) polymorphism in the KLKB1 gene, encoding plasma kallikrein, was also investigated. Carriers of the G allele of the KLKB1-428G/A polymorphism exhibited a significantly delayed disease onset [47]. Since an earlier onset of symptoms is inversely correlated with the subsequent course of the disease, these polymorphisms may be helpful prognostic biomarkers of disease severity.
This entry is adapted from the peer-reviewed paper 10.3390/jcm10092023