Calcium ions (Ca2+) play an important role as second messengers in regulating a plethora of physiological and pathological processes, including the progression of cancer. Several selective and non-selective Ca2+-permeable ion channels are implicated in mediating Ca2+ signaling in cancer cells. In this review, we are focusing on TRPC1, a member of the TRP protein superfamily and a potential modulator of store-operated Ca2+ entry (SOCE) pathways. While TRPC1 is ubiquitously expressed in most tissues, its dysregulated activity may contribute to the hallmarks of various types of cancers, including breast cancer, pancreatic cancer, glioblastoma multiforme, lung cancer, hepatic cancer, multiple myeloma, and thyroid cancer.
1. Introduction
Calcium is a ubiquitous second messenger that is known to regulate a myriad of physiological cellular functions in normal cells [
1]. Levels of intracellular free concentration of Ca
2+ is tightly regulated with strict spatial and temporal control to initiate, maintain, and terminate appropriate signaling pathways and phenotypes. Changes in intracellular Ca
2+ concentration include fast processes that require milliseconds of intracellular Ca
2+ spikes necessary for exocytosis and muscle contraction, to processes requiring minutes to hours of Ca
2+ flux that affect cellular proliferation, cell cycle control, migration, gene expression, and cell death [
2,
3]. Ion channels play a fundamental role in defining regulatory signaling pathways in the progression of cancer. Further, numerous studies indicate that Ca
2+-dependent signaling pathways are involved in augmenting tumor proliferation, differentiation, migration, invasion, metastasis, and apoptosis, thus tumor cells often exhibit increased expression of Ca
2+ regulatory networks [
2,
3,
4]. Moreover, given the importance of Ca
2+ signaling, it is not surprising that cells tightly and precisely regulate proteins handling Ca
2+ signals, including receptors, channels, and transporters [
5]. Regulation of Ca
2+ homeostasis includes transient Ca
2+ release from intracellular stores (ER/SR, mitochondria, lysosomes) as well as more sustained influx of extracellular Ca
2+ [
6]. The store-operated Ca
2+ entry pathway (SOCE) is a major Ca
2+ entry pathway in non-excitable cells and SOCE activity is known to modulate insensitivity to antigrowth signals via multiple routes, as reviewed by Prevarskaya et al. [
4]. There are numerous channels and transporters that regulate Ca
2+ levels, and deciphering the role and interplay of individual members in facilitating tumor progression remains challenging. There is a great deal of controversy surrounding the role of TRPC1; while some reports suggest it is an ion channel, other reports suggest that TRPC1 alone is not sufficient to form an ion channel and functions as a modulator of other TRPC channels. Additionally, the expression of TRPC1 as a prognostic marker in cancer appears to be context specific as TRPC1 expression has been reported to be associated with poor clinical outcomes for certain types of cancers, while in other indications it is reported to be associated with improved outcomes. This review will focus on the role of TRPC1 expression as a determinant of SOCE and cancer progression.
The transient receptor potential (TRP) ion channels were first discovered in
Drosophila melanogaster by studying photo-transduction [
7]. The TRP protein superfamily shares similarities in structure to the parent
Drosophila TRP and were initially classified into three subfamilies TRP-Canonical, TRP-Vanilloid, and TRP-Melastatin (TRPC, TRPV, and TRPM, respectively) [
8]. Later, the TRP superfamily was classified into seven subfamilies; TRP-Classical/Canonical (TRPC), TRP-Vanilloid (TRPV), TRP-Melastatin (TRPM), TRP-Ankyrin (TRPA), TRP-Polycystin (TRPP), and TRP-Mucolipin (TRPML). The non-mechanoreceptor potential C-TRP (TRPN) is comprised of approximately 30 members [
9]. Except for TRPM4 and TRPM5, which are Ca
2+-activated monovalent-selective cation channels [
10,
11], TRP family members are non-selective channels that are permeable to Ca
2+ to varying degrees [
9]. TRP channels generally share structural similarities that include six-transmembrane domains, and the proteins typically assemble as homotetrameric or in some cases heterotetrameric channels summarized by Strubing and colleagues [
12].
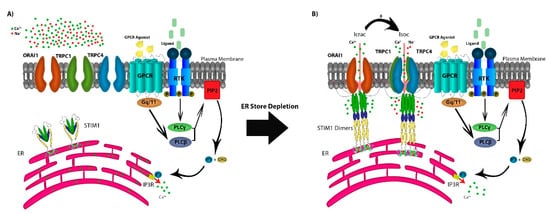
In addition to TRP channels, the SOCE mechanism of action is dependent on the depletion of the endoplasmic reticulum (ER) Ca
2+ stores through ryanodine receptors (RyRs) or inositol 1,4,5-trisphosphate receptors (IP
3R) [
13,
14]. SOCE is regulated by agonist binding surface receptors, including G-protein coupled receptors (GPCRs) or receptor tyrosine-kinases (RTKs), activating phospholipase Cβ (PLCβ) via Gq/11 and PLCγ via RTK-mediated signaling [
2,
6]. This results in the enzymatic cleavage of plasma-membrane phosphatidylinositol 4,5-bisphosphate (PIP
2) into IP
3 and diacylglycerol (DAG). The depletion of Ca
2+ stores from the ER is sensed by the transmembrane protein stromal interaction molecules (STIM1 and STIM2), as Ca
2+ dissociates from the EF domain of STIM1 and/or STIM2 [
15]. STIM molecules multimerize and translocate to ER–PM junction to form puncta that co-assemble with any or all of three calcium-release-activated calcium (CRAC) channel subunits ORAI1/2/3. This protein–protein interaction between STIM and ORAI results in the sustained opening of the highly Ca
2+-selective CRAC channels that allow for both cytosolic Ca
2+ signaling and replenishing of ER stores [
6]. Additionally, in some cell types, STIM1 may intersect with ORAI1 and members of the TRPC subfamily by its reported capability to directly interact with TRPC1, TRPC4, and TRPC5, and indirectly with TRPC3 and TRPC6 (A) [
16,
17,
18,
19].
Figure 1. The store-operated Ca2+ entry pathway (SOCE). (A) SOCE is regulated by agonist binding to G-protein coupled receptors (GPCRs) or receptor tyrosine-kinases (RTKs), activating phospholipase Cβ (PLCβ) via Gq/11 and PLCγ via RTK-mediated signaling, resulting in the production of IP3 and DAG from the cleavage of plasma-membrane PIP2. IP3 depletes Ca2+ stores from the ER through the IP3R which is sensed by STIM1. (B) STIM molecules multimerize forming puncta and translocate to the ER–PM junction, co-assembling with the CRAC channel subunits ORAI1, activating the Ca2+ selective Icrac currents. Further, STIM1 forms the STIM1-ORAI1-TRPC1 complex activating cation non-selective Isoc currents.
The TRPC subfamily consists of seven members (TRPC1-7), and they are known to function as non-selective cation channels, with permeability to Ca
2+, Na
+, and K
+ [
20]. The role of TRPC1 in SOCE activity has been discussed in a recent report by Dyrda and colleagues, where they reported that TRPC1 activation is dependent on activation of the Icrac current activated by STIM1 and comprised of ORAI1/2/3 [
21]. However, activation of STIM1 does not necessarily activate TRPC1, as there are two proposed mechanisms for the store-operated channels activation. The transmembrane protein STIM1 interacts with ORAI1 activating the CRAC channels, with Ca
2+ selective Icrac currents [
22,
23,
24,
25]. STIM1 interacts with TRPC1, forming the STIM-ORAI1-TRPC1 complex and activating the SOC channels conducting cation non-selective Isoc currents [
25,
26]. This experimental evidence supports a model in which, following the activation of the SOC channels, the non-selective cation channels TRPC1, TRPC4, and TRPC5 form heterotetramers, as TRPC1-TRPC4, TRPC1-TRPC5, or TRPC1-TRPC4-TRPC5 become operative to facilitate further Ca
2+ entry (B) [
27,
28,
29]. TRPC1 has been under study in recent years as a Ca
2+ channel in the SOCE pathway, on the other hand, recent reports have suggested that TRPC1, when expressed on it is own, is not sufficient to form a channel. The role of TRPC1 has been studied extensively in recent years to investigate its function as a calcium channel or a modulator for other TRPC channels. Despite all data and studies into TRPC proteins, their mechanism of action remains poorly understood. Some members of the TRPC subfamily are capable of forming channels when expressed alone by forming homomers (TRPC4-TRPC4 and TRPC5-TRPC5) [
30]. However, Storch et al. reported that TRPC1 is incapable of forming a Ca
2+-permeable channel by itself, but is essential in forming heteromers with all other members of the TRPC subfamily [
31]. Further, they reported a decrease in Ca
2+ permeability in heteromeric complexes containing TRPC1, but the effect of TRPC1 in Ca
2+ entry and as a member of the SOCE pathway is tissue dependent.
2. Ca2+ Signaling Through SOCE Modulates Gene Expression
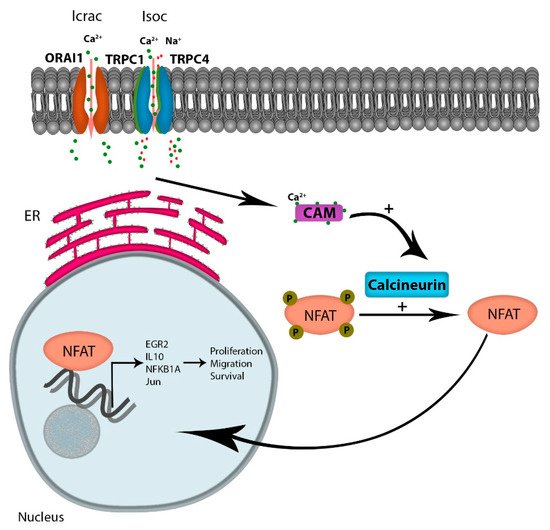
Considering the numerous pathways that Ca
2+ signaling modulates, including kinases, phosphatases, proteases, and metabolic enzymes, the role of Ca
2+ signaling in the progression of cancer is likely multi-factorial. However, one attractive Ca
2+-dependent candidate pathway is the activation of NFAT (reviewed by Putney [
35]). Increased intracellular Ca
2+ levels occur via multiple channels, but the increased oscillatory Ca
2+ currents through emptying of intracellular stores, and extracellular Ca
2+ flux through the SOCE is the major contributor to Ca
2+ -regulated gene expressions [
36]. Ca
2+ binding to its receptor calmodulin activates the phosphatase protein calcineurin, which in turn dephosphorylates NFAT, leading to its translocation to the nucleus. NFAT is a transcription factor known to regulate the expression of genes encoding cytokines and receptors mandatory for T-cell survival. Multiple reports indicate that tumor cells have hijacked the calcium-dependent NFAT pathway to support cytokine-dependent survival and homing. For example, in diffuse large B-cell lymphoma (DLCBCL), Bucher et al. reported that NFAT activity is chronically elevated in tumor cells; a finding that correlated with elevated Ca
2+ levels. In addition, inhibition of calcineurin with cyclosporin A or FK506 treatment reduced NFAT target genes including EGR2, IL10, NFKB1A, and Jun, and induced cell death in DLCBCL cell lines [
37]. Similarly, Urso et al. showed that NFATc3 is a critical determinant of proliferation and migration of U251 glioblastoma cell lines [
38]. The authors demonstrated that reducing the expression of NFAT3c inhibited the expression of TNF-alpha, GM-CSF, IL-2, and CXCR-3 when U251 cells were treated with an ionophore. Finally, the authors demonstrated that a reduction of NFAT3c inhibited the growth of U251 cells in vivo. The role of NFAT in cancer progression has been comprehensively reviewed by Mancini et al., where they reported that NFAT overexpression in solid and hematological tumors plays a role in tumor survival, migration, and invasion [
39] ().
Figure 2. Ca2+ entry through SOCE activates NFAT activation: Ca2+ entry through the Icrac channel binds calmodulin, leading to the activation of the phosphatase protein calcineurin, activating the transcription factor NFAT. Active NFAT is translocated to the nucleus, regulating the expression of genes promoting proliferation, migration, and survival.
3. TRPC1 Expression and Correlation With Proliferation, EMT, and Migration
3.1. Pancreatic Cancer
Epithelial-mesenchymal transition (EMT) is a known modulator and a key step into tumor invasion and metastasis. One of the key modulators of EMT is TGF-β, which has been reported previously to induce EMT in mammary epithelial cells [
65]. The role of TRPC1 as a Ca
2+ channel in pancreatic cancer cell proliferation and its development was proposed as being a downstream effector of TGFβ signaling [
52,
56]. In SMAD4-null pancreatic cancer cells, TGFβ is reported to induce a cytosolic Ca
2+ increase, leading to the activation of the Ca
2+-dependent protein kinase C-α (PKCα) and its translocation to the plasma membrane. Further, TGFβ activated PKCα-dependent cellular motility and migration, by inhibiting the tumor suppressor PTEN [
56]. Later, TRPC1 4 and 6 levels were shown to be high in pancreatic cancer cells, indicating their function as TGFβ mediators for Ca
2+ entry [
52]. Pharmacologically inhibiting SOCE pathways using 2-APB and La
3+ abrogated the TGFβ-dependent increase in cytosolic Ca
2+ levels. Blocking PKCα by the selective PKCα inhibitor Gö-6976 also inhibited TGFβ-induced Ca
2+ entry [
52]. Further, siRNA knockdown of TRPC1 or treatment with 2-APB significantly inhibited pancreatic cancer cell motility induced by TGFβ, although, interestingly, siRNA knockdown of TRPC4 and 6 had no effect on TGFβ-induced pancreatic cell motility [
52].
3.2. Breast Cancer Epithelial-Mesenchymal Transition and Proliferation
It has previously been reported that epithelial–mesenchymal transition (EMT) is associated with upregulation of SOCE via increased levels of STIM1 and ORAI1 in MCF7 and MDA-MB-231 cell lines, promoting invasion and proliferation of breast cancer cells [
54]. However, the role of the SOCE pathway in EMT may be dependent on the stimulus inducing the transition and/or cell context specific. For example, MDA-MB-468 cell lines undergoing epidermal growth factor (EGF)-induced EMT correlate with a reduction of SOCE activity, and the reduction in Ca
2+ flux was associated with ORAI1 downregulation, while TRPC1 expression was not altered [
50]. Interestingly, hypoxia-induced EMT increased the expression of TRPC1 in MDA-MB-468, MDA-MB-231, and HCC1569 cell lines, an effect that required HIF1α expression, indicating that TRPC1 is a HIF1α target. Reducing TRPC1 expression inhibited hypoxia-induced increased Snail, Vimentin, and Twist expression. Reducing the expression of TRPC1 in hypoxia resulted in decreased basal Ca
2+ levels, but increased SOCE activity was noted by depleting intracellular stores using sarco/endoplasmic reticulum Ca
2+ ATPase (SERCA) inhibitor cyclopiazonic acid [
42]. Reducing the expression of TRPC1 in MDA-MB-468 cells decreased the proliferation rate and was associated with reduction of cells in the S-phase, while ORAI1 had no effect [
50].
MCF7 breast cancer cells proliferate in response to activation of the Ca
2+-sensing receptor (CaR) by extracellular Ca
2+ or its agonist spermine [
66]. El Hiani and colleagues reported that Ca
2+-mediated CaR activation in MCF7 cells results in activation of PLC and PKC [
55]. They further reported that proliferation is dependent on activation of ERK1/2, which was shown to be activated downstream of PLC and PKC. Reducing the expression of TRPC1 attenuated ERK1/2 phosphorylation mediated by CaR activation, which is necessary for CaR-induced cell proliferation of MCF7 cells. In addition, reducing TRPC1 expression inhibited MCF7 proliferation by halting the cell cycle progression at the G1 phase [
45]. Cell cycle progression was dependent on TRPC1 mediating the activity of Ca
2+-activated K
+ channels (KCa3.1) [
45,
55]. Interestingly, a feed-forward loop was described where TRPC1 expression was dependent on activation of EGFR and ERK activity in MCF7 cells [
67]. In human breast ductal adenocarcinoma primary patient samples, TRPC1, TRPC6, TRPM7, and TRPM8 were reported to be overexpressed in cancer cells compared to normal adjacent tissue. Importantly, increased TRPC1 expression correlated with expression and increased proliferative and invasive capacity of small grade I breast cancer tumors [
68]. Taken together, these data suggest that TRPC1 expression may play a role in facilitating EMT and proliferation, and further studies are required to carefully delineate the mechanism underpinning the role of TRPC1 in mediating EMT and proliferation in breast cancer.
3.3. Glioblastoma
In D54MG cells, a model for malignant gliomas or glioblastoma multiforme (GBM), the pharmacological inhibition of SOCE with 2-APB, SKF96365, and MRS1845 significantly reduced both Ca
2+ influx and proliferation as well as the formation of multinucleated cells, a characteristic of GBM [
53]. To define the role of TRPC1 as a Ca
2+ channel involved in SOCE, its function was blocked using a polyclonal-TRPC1 antibody, which reduced the Ca
2+ entry by 25%, whereas no effect was seen with TRPC5 inhibition [
53]. Similarly, Ca
2+ entry was significantly decreased following the reduction of TRPC1 expression using shRNA [
53]. The knockdown of TRPC1 also significantly reduced proliferation and resulted in enlarged multinucleated cells. Moreover, TRPC1 expression levels were reportedly down-regulated in patients with giant cell glioblastoma [
53].
It has been reported that lipid rafts microdomains (LRDs) and plasma membrane caveolin-1 are critical for TRPC1 insertion into the plasma membrane and its activity as a Ca
2+ channel, as TRPC channels contain a caveolin-1 binding domain [
69,
70,
71]. Indeed, TRPC1 was implicated in chemotaxis and directional migration of D54MG cells towards epidermal growth factor (EGF), by co-localizing with caveolin-1 and lipid rafts in the cells’ leading edge, which was abrogated with inducible TRPC1 shRNA knockdown [
51]. Consistent with the importance of Ca
2+ signaling, the use of SOCE inhibitors MRS1845 and SKF96365 disrupted glioma migration [
51]. Evidence that TRPC1 expression is essential for growth in vivo was assessed by injecting nude mice glioma cells ectopically expressing doxycycline-inducible shRNA targeting TRPC1. Mice with inducible knockdown of TRPC1 had a shallow, significantly smaller tumor size compared to wild-type TRPC1 expression. However, propensity for metastasis was not evaluated in these mice [
53].
3.4. Lung Cancer
Lung cancer is the leading cause of death from cancer in men and women and thus clinical data continue to indicate the need for new treatment strategies to improve patient outcomes [
72]. Levels of TRPC protein expression and their relation to tumor prognosis have been reported previously and identified as a potential target for treatment. TRPC1, TRPC3, TRPC4, and TRPC6 levels have been found to be highly expressed in patient specimens compared to other TRPC channels [
49,
73]. Moreover, overexpressing TRPC1 and TRPC6 in the A549 NSCLC cell line was sufficient to increase proliferation, while blocking TRPC channels by the IP
3 receptor inhibitor and SOCE modulator 2-APB, or the specific TRPC1 antibody T1E3, inhibited proliferation of A549 cells [
49].
As mentioned earlier, TRPC1 levels in breast cancer were dependent on HIF-1α following hypoxia-induced EMT. Wang et al. reported similar results in lung cancer by exposing A549 cells to high nicotine levels, which in turn resulted in increased HIF-1α levels (as shown previously by Guo et al. [
74]), leading to the upregulation of SOCE components, namely ORAI1, TRPC1, and TRPC6 [
75]. Further, the nicotine exposure was associated with increased basal intracellular Ca
2+ levels in A549 cells related to constitutive SOCE activity. Downregulating HIF-1α was associated with low expression of ORAI1, TRPC1, and TRPC6, and resulted in decreased proliferation of A549 cells. Silencing TRPC1 decreased hypoxia-induced autophagy [
75]. Autophagy is known to promote survival in hypoxic environments [
76], albeit no data was reported on survival with respect to exposure to hypoxia and TRPC1 expression. More recently, STIM1 and TRPC1 were shown to mediate cisplatin cytotoxicity in NSCLC A549 cell line by facilitating the DNA damage response (DDR) and reactive oxygen species (ROS) production leading to apoptosis. While these effects were discovered mostly by silencing STIM1, and thus do not rule out ORAI1 or other SOCE components, the overall effect was mainly mediated by inhibition of Ca
2+ influx through the SOCE pathway [
40].
3.5. Colon Cancer
In colon cancer, the role of SOCE members, mainly TRPC1 and ORAI1, has been discussed thoroughly by Villalobos et al. [
77]. Basal Ca
2+ levels have been shown to be higher in HT29 colon carcinoma cell lines with higher increase in cytosolic Ca
2+ in response to agonists like ATP and carbachol, compared to the normal human mucosa cell line NCM460 [
48]. Although TRPC1 mRNA levels were similar to other SOCE members in HT29 cells, there was an increase in protein expression levels compared to others, and TRPC1 silencing was associated with decreased store-operated (Isoc) currents. As previously mentioned, TRPC1 facilitates the migration of GBM cells towards EGF. Guéguinou et al. investigated the mechanism by which TRPC1 contributes to colon cancer cell migration. They demonstrate that reducing the expression of TRPC1 inhibits migration of the HCT-116 colon cancer cell line by disrupting the complex formation of Ca
2+-activated K
+ channels (SK3), ORAI1, and TRPC1 in the lipid rafts [
44]. Further, colon cancer cell migration was shown to be dependent on EGFR activation, leading to downstream activation of the PI3K/Akt pathway. This promotes the phosphorylation activation of STIM1 and SOCE, leading to the translocation of TRPC1 and ORAI1 in the lipid rafts to form a complex with SK3, which allows a loop formation of further Akt activation and TRPC1-ORA1-SK3-dependent migration. Taken together, TRPC1 expression appears to have a role in the migration of multiple cancer types and more studies are required to determine whether inhibition of this pathway will block metastasis of primary tumors.
Table 1. Role of TRPC expression or activity in augmenting proliferation and metastasis in cancer.
| Cancer Type |
Cell Type |
Native TRPC Expression |
Silenced Proteins/ Tools Used |
Native Expression Effect /Silencing Effect |
Reference |
| Pancreatic |
BxPc3 (human ductal adenocarcinoma) |
↑ TRPC1 |
|
Motility/ ↓ motility (TGFβ-dependent motility) |
[52] |
| Breast |
MDA-MB-468 (EGF-mediated EMT cells) (human breast adenocarcinoma) |
Comparable to MDA-MB-231 - EMT |
TRPC1/siRNA |
/↓ Cell proliferation (↓S-phase) |
[50] |
MDA-MB-468
hypoxia-mediated EMT cells) |
↑ TRPC1 and TRPC3 |
TRPC1/siRNA |
/↑ Ca2+ influx in SOCE and ↓ autophagy marker LC3BIII |
[42] |
| MCF7 (adenocarcinoma) |
↑TRPC1 |
TRPC1/siRNA |
Proliferation/↓ proliferation (↓G1-phase) |
[45,55] |
| Primary patient TNBC cells (mesenchymal subtype) |
↑ TRPC1 |
- |
Worsened prognosis/ - |
[42] |
| Primary human breast ductal adenocarcinoma |
↑ TRPC1
↑ TRPC6 |
- |
↑ proliferation and invasion/- |
[68] |
| Glioblastoma Multiforme |
D54MG (GMB Cell line) |
- |
TRPC1/2-APB, SKF96365, MRS1845, polyclonal TRPC1 antibody, and shRNA |
Proliferation, migration/ ↓ Ca2+ influx in SOCE, ↓ proliferation |
[51,53] |
| Lung |
Primary patient cells (NSCLC) |
↑ TRPC1, 3,4,6 |
- |
High expression with well-differentiated tumor |
[49,73] |
| A549 (NSCLC cell line) |
↑ TRPC1
↑ TRPC6 |
|
↑ Proliferation/↓ proliferation |
[49] |
| A549 (hypoxia-mediated EMT by nicotine treatment) |
↑TRPC1
↑TRPC6,
and ↑ORAI1 |
↓TRPC1/siRNA HIF-1α |
↑ SOCE activity/↓ proliferation, ↓hypoxia-induced autophagy |
[75] |
| Colon |
HT29 (human colon carcinoma) |
↑ TRPC1 (protein) |
|
↑ SOCE, ↑ proliferation/↓ Isoc currents, ↓ invasion |
[48] |
| |
HCT116 |
- |
TRPC/siRNA |
Migration/↓ migration |
[44] |
This entry is adapted from the peer-reviewed paper 10.3390/cells9020388