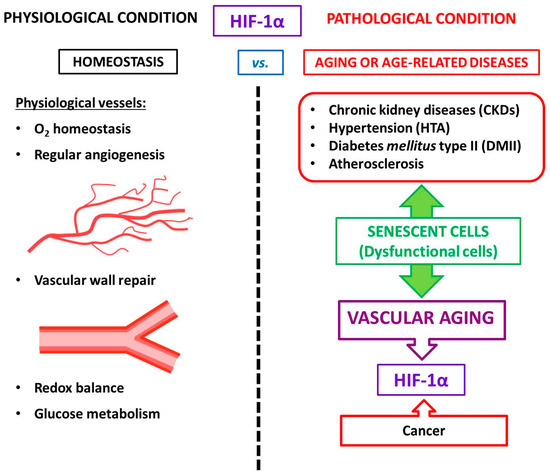
According to current knowledge, the age-dependent impairment of HIF-1α induction leads to diminished vascular responses to limb ischemia [
54] and less effective wound healing [
81]. Some evidence shows the functionally important expression of HIF-1α among ischemic limb mice. It has been demonstrated that the abundance of the HIF-1α protein is decreased in ischemic tissues from aged mice and has also been linked with the downregulation of genes encoding angiogenic growth factors [
61]. In this regard, Bosch-Mache et al. showed that reduced blood flow recovery among aged mice resembles the response of heterozygous HIF-1α knockout mice to ischemia [
54]. Interestingly, the exogenous administration of constitutively active forms of HIF-1 into the ischemic limb was sufficient for overcoming the age-dependent impairment of ischemia-induced vascular remodeling in aged mice [
54]. Thus, the ability of HIF signaling to regulate the angiogenic process may be one of the main factors in vascular aging. In this scenario, the HIF pill theory would provide a preventive treatment for vascular aging.
Some aging-related vascular diseases present a rapid course of regular age-dependent arterial changes called early vascular aging (EVA) [
82]. Aging of the arterial wall vessels in humans can be quantified by measuring the pulse wave velocity along the aorta—the largest elastic artery—which represents a marker of arterial stiffness [
82]. Superior techniques to evaluate EVA are the use of noninvasive procedures to determine arterial stiffness indexes, including the carotid intima–media thickness (IMT), central blood pressure, and endothelial damage parameters [
82]. In this regard, EVA is also characterized by media vascular calcification (VC) in CKD. During chronic inflammation mediated by uremic toxins, VSMCs are significantly affected, becoming dysfunctional and causing VC, which may potentially be used as a biomarker for vascular age [
16,
83,
84].
Aging is associated with transforming growth factor type β (TGF-β) inhibition via HIF-1 [
85]. In addition to TGF-β signaling, there is crosstalk between the HIF pathway and well-known stress-related sensors, including AMPK (AMP-activated protein kinase), sirtuins, and nuclear factor-κB (NF-κB) [
86]. Despite the role of sirtuins in the regulation of aging and longevity, nowadays, its role is still controversial [
87]. Satoh et al. [
87] described that sirtuin-1 activity is critical in the systemic regulation of tissue communication, aging, and longevity in mammals. Notably, overexpression of sirtuin-1 due to the calorie restriction diet plays a role in the pathogenesis of age-associated mitochondrial damage in aging kidney mice [
88]. Recently, it has been demonstrated that sirtuin-1 and HIF-1α are connected [
89]. In this sense, sirtuin-1 induced deacetylation of HIF-1α in aged kidneys, protecting tubulointerstitial damage [
89]. Since 2012, it is well known that sirtuin-1 is necessary for HIF-1α protein accumulation [
90] and the current knowledge is that sirtuin-1 could activate several transcriptional factors, such as HIF-1α, resulting in ameliorated mitochondria biogenesis and an extended lifespan [
91]. In the pathophysiology of vascular aging and atherosclerosis, sirtuin-1 plays a protective role [
92]. In endothelial dysfunction, the expression of sirtuin-1 is reduced promoting the manifestation of senescence in endothelial cells [
93,
94,
95]. Moreover, sirtuin-1 modulates eNOS and NO production in vascular walls [
92], playing a crucial role in maintaining vascular function and homeostasis.
Another vital player of vascular aging, which is positively regulated by HIF-1, is vascular endothelial growth factor (VEGF), a central mediator of angiogenesis. During aging, there is a defect in HIF-1 activity, yielding VEGF expression reduction and leading to the impairment of angiogenesis in response to the ischemia model [
96]. Similarly, EPO is directly regulated by HIF, and lower secretions of EPO have been observed among old animals [
97] and elderly patients [
98]. Furthermore, the transcriptional program controlled by HIF-1 includes genes involved in many aspects of cellular homeostasis, and HIF-1 abolishment by aging could generate defects in the physiological responses to hypoxia [
96]. Recently, we found that HIF-1α is involved in p53, p16, cyclin D1, and lamin B1-mediated senescence in ECs [
29]. Moreover, senescent ECs failed to express HIF-1α, and the microvesicles (MVs, an EVs subtype) released by these cells were unable to carry HIF-1α [
29]. In another study, HIF-1α was found to play a critical regulatory role in vascular inflammation among macrophages after intimal injury through limiting excessive vascular remodeling. The mechanism by which macrophage-derived HIF-1α mediated this effect is still unknown [
99]. Considering these findings, HIF-1α may represent a possible therapeutic target in vascular diseases, especially in vascular aging.
4. HIF and Atherosclerosis
Although atherosclerosis has been considered chronic inflammation, intensive research in recent years has shown that it can also be considered an age-related pathology [
28,
100]. Many pieces of evidence have demonstrated the role of vascular senescence in atherogenesis [
25,
101]. We briefly mentioned above that senolytic drugs (anti-senescence) have been proposed as a therapeutic option for cellular aging and for treating human atherosclerosis [
25,
28]. However, although gerontologists have affirmed that atherosclerosis is associated with the characteristic features of aging in humans, cardiologists believe that aging is not a risk factor for atherosclerosis. This controversial subject was re-evaluated by Minamino
et al., who demonstrated that senescent vascular cells accumulate in human atheroma and that vascular cells present features of dysfunction [
102,
103]. These and other findings suggest that cellular senescence contributes to atherosclerosis, which is a characteristic of aging in humans.
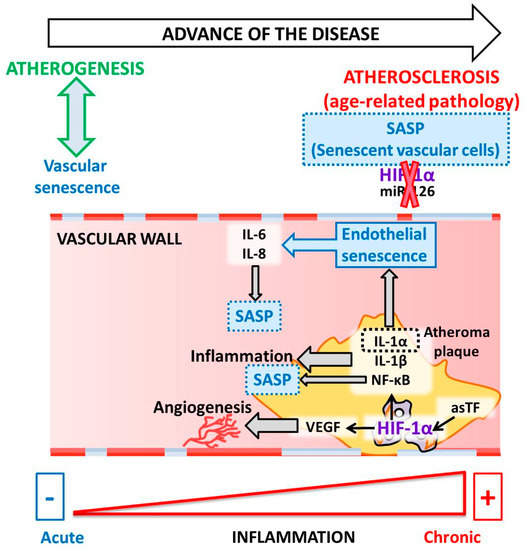
As a model for premature aging disorder, atherosclerosis is the most common type of vascular aging where the cell vessels are susceptible to damage. Adding to the complex scenario for atherosclerosis, many studies suggest that ECs and VSMCs change and acquire features of senescent cells [
104]. Moreover, during aging, blood vessels experience changes in compliance and release pro-inflammatory factors that promote atherosclerosis. Aging is associated with chronic low-grade inflammation that affects vascular and endothelial cells within the vascular wall during atherosclerosis. It is reasonable to believe that low-grade systemic inflammation may facilitate the senescent phenotype of ECs, which also contributes to the local inflammatory environment by SASP. These aging endothelium walls impair angiogenesis and decrease coagulation activity [
104]. In an aged endothelium, senescent ECs failed to achieve HIF-1α stabilization and decreased miR-126 levels, which are both essential contributors to the maintenance of endothelium homeostasis [
29].
In the vasculature, HIF-1α regulates pressure changes due to the negative regulation of TGF-β in ECs [
85] (note that pressure overload leads to increased myocardial O
2 consumption). In this regard, the anoxemia theory is defined as a condition of abnormal oxygenation of the arterial blood. This theory proposes that an imbalance between the demand for and supply of O
2 in the arterial wall is a critical factor in the development of atherosclerosis [
105]. As a consequence, macrophages become apoptotic, a necrotic core is built, and there is an eventual increase in angiogenesis, linking senescent cells to atherosclerosis progression [
7]. Therefore, the anoxemia theory is postulated to explain the progress of atheroma plaque.
In 2007, the presence of HIF-1α was described in atheroma plaque [
106]. HIF-1α is a regulator of angiogenesis and inflammation in atherosclerotic plaque destabilization. Moreover, HIF-1α is associated with an increase in VEGF levels during the inflammatory process in atheroma plaque. Notably, activated macrophages in atherosclerosis have been observed to stabilize HIF-1α under normoxic conditions [
106]. HIF-1α stabilization occurs due to the local relative hypoxia resulting from insufficient O
2 diffusion in the thickened intima and increased O
2 demand due to the local inflammatory response. If the O
2 supply is restored, HIF-1α is degraded, which reduces VEGF production and subsequent angiogenic signaling [
107]. Another study reported that HIF-1α increases as a consequence of neovascularization in complicated human atherosclerosis among human carotids, as well as in coronary plaques [
105]. Mechanistically, the angiogenic effect of the alternatively spliced tissue factor (asTF) activates HIF-1/VEGF signaling [
41]. Indeed, activated macrophages localized in atheroma plaques expressed HIF1-α and VEGF, confirming that both are critical to the regulation of human plaque angiogenesis and lesion progression. Therefore, HIF-1α mediates inflammation by promoting pro-inflammatory cytokine expression and, consequently, inflammatory cell recruitment [
108,
109]. In macrophages, HIF-1α regulates the expression of one of the major pro-inflammatory cytokines, IL-1β [
110]. Pro-inflammatory and pro-angiogenic activities are induced in endothelial cells exposed to IL-1β stimulation [
111]. Notably, anti-inflammatory IL-1β therapy led to a significantly lower rate of recurrent cardiovascular events [
112]. Another IL-1 family member, IL-1α, is mainly associated with endothelial cell senescence and atherosclerosis [
113]. Both IL-1α and IL-1β are minor components of SASP; however, these two cytokines are essential for boosting IL-6 and IL-8, which are secreted in large quantities by senescent endothelial cells [
114,
115] ().
Figure 2. Molecular pathways linking HIF-1 to senescence during atherosclerosis.
Moreover, extensive crosstalk between HIF and two master regulators of the inflammatory response, NF-κB and the signal transducer and activator of transcription 3 (STAT3), has been reported [
108,
116,
117]. Under hypoxic conditions, canonical NF-κB signaling activates HIF-1α through the interactions between p50 and p65 subunits and responsive elements in the promoter of the HIF-1α gene [
118]. The crosstalk between HIF-1α and NF-κB signaling during senescence has been investigated in several contexts, and its role in orchestrating SASP is widely accepted in atherosclerotic plaques. However, the full mechanism of this crosstalk is not yet completely understood.
Strikingly, Minamino et al. [
104] demonstrated the presence of senescent vascular cells in human atherosclerotic lesions but not in non-atherosclerotic lesions. This study characterized some of the features of senescent cells and described an increase in pro-inflammatory mediators, including NF-κB signaling-dependent mediators, but a decrease in eNOS. In addition to these findings, signs of cellular senescence have also been detected in premature aging mouse models [
119]. Together, these results provide in vivo evidence that links cellular senescence to organismal aging [
104]. For instance, arterial remodeling during atherosclerosis progression is accelerated during aging. The accumulation of lipids in the arterial wall, followed by foam cell formation, is a response to endothelial damage and inflammation. Once the atheroma plaque is stable, it becomes an advanced plaque with increased susceptibility to rupture, leading to thrombosis [
120].
Moreover, unstable plaque highlights the relationship between atherosclerosis and HIF-1α in ECs [
121] and macrophages [
122], independent of their origin (SASP or SIPS). It was also shown that the final step in atheroma plaque development is VC formation [
7,
123,
124]. Aging causes calcification in vascular smooth muscle cells, which occurs independent of inflammation but causes arterial stiffening [
120]. In this way, atherosclerosis, as well as aging or age-related atherosclerosis, causes vascular wall senescence and, as a consequence, VC—the final step of the pathology process. Increased VC in atherosclerosis produces numerous marked vascular effects, such as a reduction of tissue perfusion, which eventually causes end-organ damage, particularly in the elderly population [
125].
EVs are essential modulators of vascular cell functions relevant to vascular inflammation and atherosclerosis [
126,
127]. Furthermore, EVs have been identified as cell-to-cell communicators. EV content includes proteins, lipids, and nucleic acids that are transferred to target cells [
15,
128] and modulate cell functions and phenotypes [
129]. The abundance of EVs and the release of their cargos are augmented under inflammatory [
129] and pathological conditions, including CVD, metabolic disorders, atherosclerosis, and DMII [
129,
130]. Platelets liberate EVs from vascular vessels (ECs and smooth muscle cells), erythrocytes, and leukocytes [
131,
132]. EVs may be potential biomarkers and pharmacological targets for atherosclerotic diseases and, therefore, may also be biomarkers for age-associated diseases, especially for EV-based therapeutics.