Multimeric PET radioligands consist of identical binding motifs (pharmacophores) connected to a single backbone (linker) attached to a group, which can be labeled with a positron-emitting radionuclide suitable for PET molecular imaging (radiolabeled domain).
- PET imaging
- multimeric radioligands
- integrin αvβ3
- cyclic RGD
1. Introduction
The idea of multimeric radioligand, which consists of multiple structural motifs connected to a single backbone, started from the need to achieve higher binding avidity through cooperative multivalent interactions. In a way, this mimics the behavior of antibodies, which have multiple individual weak interactions with the target. These multiple weak interactions combine in a cooperative way and increase binding avidity [1][2]. Several binding modes have been proposed as an explanation for this cumulative effect, such as the simultaneous binding of the ligand to multiple receptors on the cell surface (Figure 1a), or the improved statistical effect, where the ligand binds to one receptor, but the ligand’s apparent local concentration is increased (Figure 1b) [1].
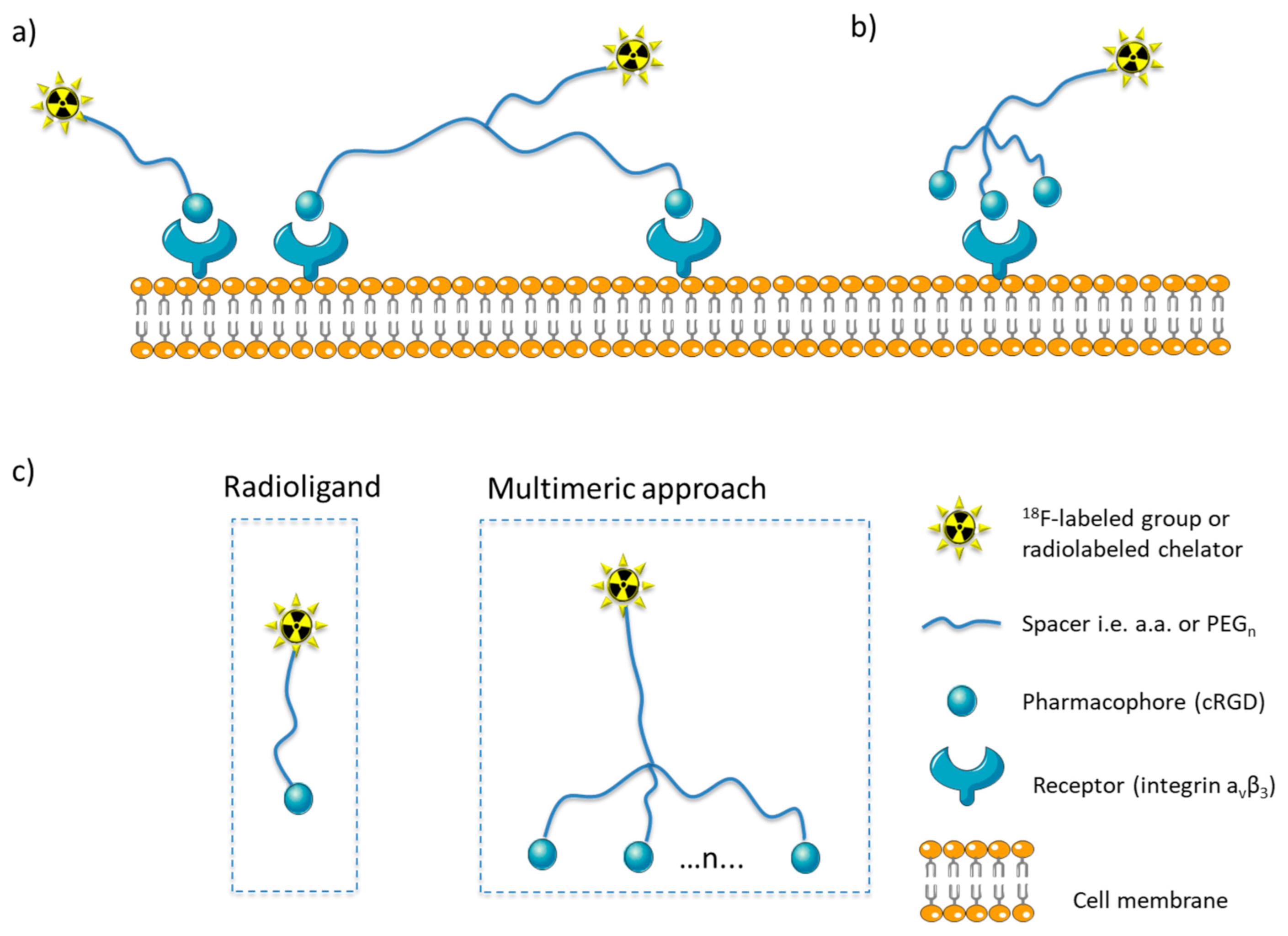
Figure 1. Binding models for multimers on the cell surface: (a) The binding of a radioligand to a cell surface receptors and the multimeric approach resulting in simultaneous binding of two pharmacophores connected via a long linker with two receptors, (b) improved binding efficiency of a ligand, due to the increased apparent local concentration of the pharmacophore (statistical effect) in the micro-environment of the receptor; (c) basic principles for the design of monomeric and multimeric radioligands (where n = number of pharmacophores).
Multimeric radioligands resemble a typical radioligand by retaining some of its structural features—e.g., a chelator group for radiometals—and differ from it, as they include multiple pharmacophores (Figure 1c). Radioligands targeting specific receptors on tumor cell surfaces consist of a receptor binding section (pharmacophore or ligand), and a radiolabeled domain. The latter is either a radiometal chelator, which coordinates with a radiometal, or a suitable group for radio-halogenation, which is often connected with the pharmacophore through a linker (spacer) group. Linker groups are used (Figure 1): (1) to increase the distance between the ligand and the chelator group and thus, minimize the interference of the radiolabeled domain with the binding pocket of the ligand in the receptor; (2) to enhance the interactions between the ligand and its binding pocket; (3) to enable the addition of a second or of multiple binding pharmacophore groups leading to multimeric ligands; (4) to increase the distance between the multiple pharmacophores in order to enable simultaneous binding on more than one receptors (Figure 1a); (5) to change the pharmacokinetic properties of the radioligand, altering its hydrophobicity and eventually increase its apparent local concentration when it reaches the receptor’s binding area [3][4][5].
The linker properties are very important since they influence the orientation of the ligand, when it approaches the extracellular receptors binding domain, enabling or preventing simultaneous multi-receptor binding of the multimers [6]. Long, flexible linkers increase the distance between the binding domains, permitting simultaneous binding of the ligand to multiple receptors, while rigid linkers have less entropy penalty from binding [1].
For multimeric radioligands, a wide spectrum of pharmacophores has been used like small molecules, peptides, or fractions of antibodies [1][6][7][8][9]. A limited number (n = 2–8) of pharmacophores e.g., c(RGD)n has been covalently connected using low molecular weight linkers e.g., polyethylene glycols (PEGs), while higher numbers of pharmacophores have been tethered on the surface of large molecules, such as liposomes or nanoparticles.
2. PET Diagnostic Molecules Utilizing Multimeric Cyclic RGD Peptide Analogs for Imaging Integrin αvβ3 Receptors
Multimeric PET radioligands consist of identical binding motifs (pharmacophores) connected to a single backbone (linker) attached to a group, which can be labeled with a positron-emitting radionuclide suitable for PET molecular imaging (radiolabeled domain). Among the various PET multimeric radioligands investigated for targets like integrin αvβ3, PSMA, GRPr, VEGFR, and EGFR-TKI, the ones targeting integrin αvβ3 are the most studied and the only category which has reached the clinical stage of development (Table 1). Multimeric c(RGD) analogs are a fine example, providing proof that multimerization can improve ligands characteristics like receptor avidity and tumor uptake.
Several factors regarding the design of c(RGD) multimeric radioligands should be taken under consideration. One of the most important factors is the length and flexibility of the linker (1) connecting the chelator group with the multimeric scaffold and (2) connecting the various pharmacophores. Regarding the first case of linker (1), several examples of 18F-labeled compounds, 7–10 have shown that the introduction of PEG group in-between the pharmacophores and the labeling site not only improves the overall radiolabeling yield but also reduces the renal uptake and increases tumor-targeting efficacy [10]. Specifically, the introduction of PEG3 minimized the instability factors observed in the acidic and high-temperature radiolabeling conditions of 10, due to the thiourea linkage of the labeling site with the α-amine of the Glu linker [11]. Considering the second case (2), the length of the linker defines the distance between the two pharmacophores. Cyclic RGD dimeric peptides, where the Glu linker was connected with each c(RGD) using additional groups like PEG4 or G3 e.g., 14–16 or 29–32 have shown better results, with respect to tumor uptake, than other ligands with shorter linkers [12]. In particular, for c(RGD) dimers, it seems that this distance has to be in the range of 30–38 bonds like in 26, 27 [13] or the SAR conjugate 33 [14][15] for achieving bivalency, and eventually leading to higher integrin αvβ3 binding avidity [12].
Several pharmacodynamic models have been proposed as an explanation for the observed improvements in binding avidity, reduced receptor off-rate, which eventually result in high tumor uptake. One of the models suggests simultaneous binding of the ligand with two receptors on the cell surface; this can be accomplished with the utilization of extremely long spacers, which cover the distance between two receptors on the cell surface. However, extremely long spacers do not always prove to be advantageous for the pharmacodynamic or pharmacokinetic ligand characteristics, because they may prevent other actions such as the internalization of the ligand or may worsen its pharmacokinetic properties, resulting in reduced tumor uptake. Another model describing the improved effects observed for multimeric ligands is based on the improved statistical effect, in this case, the ligand binds to one receptor, but its apparent local concentration of the ligand in proximity to the receptors is increased. This seems to be the most likely explanation for ligands with short linkers [1].
Another factor regarding the design of c(RGD) multimers is the number of c(RGD) pharmacophores included. Increasing the number of pharmacophores had a positive effect on ligand avidity and cell binding in vitro. Nevertheless, this effect did not always correspond with similar advantages in vivo, since increasing peptide multiplicity, in many cases resulted in a parallel increase of ligand uptake in normal organs. Thus, the benefits of multiplicity seem to have limits. Among the multimers summarized in this article, the dimers seem the most successful cases and that is also the reason they have further advanced in the clinic, over the other tracers.
Several examples of c(RGD) multimers for PET imaging of integrin αvβ3 have been studied so far, which have been labeled with various PET radionuclides i.e., 18F, 68Ga, and 64Cu. The 18F analogs and specifically dimeric [18F]F-c(RGD)2 ligands have been the most successful and have already reached the stage of clinical development for various applications utilizing integrin αvβ3 imaging like cancer (ligands 8, 10, and 14). However, the NOTA analogs, which are labeled using [18F]AlF, a much easier, faster, and high yielding production procedure (40 min and 20 min for kit radiolabeling, yield 42%, radiochemical purity > 95%), have significant advantages for their future clinical application, especially after the introduction of kit formulations. Among the two compounds developed so far: [18F]Alfatide I 10 and [18F]Alfatide II 14; the second is considered more stable regarding a possible intermolecular Lewis acid-catalyzed hydrolysis during its production. Besides 18F, another popular positron-emitting radioisotope is 68Ga, which can be produced by a 68Ge/68Ga generator. So far one clinical study has been published for each of the 68Ga labeled dimeric structures, 12 and 15. Tracer 12 showed similar pharmacokinetics with 10, which can also be labeled with 68Ga, and is of alike chemical structure to 14. Consequently, additional results of clinical studies regarding [68Ga]Ga-c(RGD)2 dimers i.e., 12 or 15 are expected to be published in the near future.
Finally, regarding the advantages of multimeric c(RGD) analogs in clinical studies, according to the limited data generated so far, the dimeric structures seem to have an advantage over their monomeric analogs. Even so, additional evaluation is needed with large cohorts of patients in order to determine if the multimeric strategy provides higher sensitivity and specificity for tumor detection and staging over the monomeric RGD compounds and the golden standard [18F]FDG.
3. Concluding Remarks
Multimerization is generally advantageous and multimeric c(RGD) radioligands have increased binding avidity against αvβ3 receptors and are more effective in tumor targeting compared to monomeric c(RDG) radioligands. However, when the number of c(RGD) pharmacophores was increased above 2, it did not enhance the pharmacokinetic properties of the ligand in vivo, despite the benefits of multimerization observed in vitro. The length and flexibility of the linker connecting the c(RGD) pharmacophores and the linker connecting the multimerization scaffold with the chelator group have a significant role in the biological activity of the multimeric c(RGD) ligands. Clinical studies are expected to bring forward valuable information regarding the application benefits of multimeric c(RGD) ligands.
This entry is adapted from the peer-reviewed paper 10.3390/molecules26061792
References
- Handl, H.L.; Vagner, J.; Han, H.; Mash, E.; Hruby, V.J.; Gillies, R.J. Hitting multiple targets with multimeric ligands. Expert Opin. Ther. Targets. 2004, 8, 565–586.
- Crothers, D.M.; Metzger, H. The influence of polyvalency on the binding properties of antibodies. Immunochemistry 1972, 9, 341–357.
- Yim, C.-B.; Van Der Wildt, B.; Dijkgraaf, I.; Joosten, L.; Eek, A.; Versluis, C.; Rijkers, D.T.S.; Boerman, O.C.; Liskamp, R.M.J. Spacer Effects on in vivo Properties of DOTA-Conjugated Dimeric [Tyr3]Octreotate Peptides Synthesized by a “CuI-Click” and “Sulfo-Click” Ligation Method. ChemBioChem 2011, 12, 750–760.
- Baranski, A.-C.; Schäfer, M.; Bauder-Wüst, U.; Wacker, A.; Schmidt, J.; Liolios, C.; Mier, W.; Haberkorn, U.; Eisenhut, M.; Kopka, K.; et al. Improving the Imaging Contrast of 68Ga-PSMA-11 by Targeted Linker Design: Charged Spacer Moieties Enhance the Pharmacokinetic Properties. Bioconjugate Chem. 2017, 28, 2485–2492.
- Liolios, C.C.; Fragogeorgi, E.A.; Zikos, C.; Loudos, G.; Xanthopoulos, S.; Bouziotis, P.; Paravatou-Petsotas, M.; Livaniou, E.; Varvarigou, A.D.; Sivolapenko, G.B. Structural modifications of 99mTc-labelled bombesin-like peptides for optimizing pharmacokinetics in prostate tumor targeting. Int. J. Pharm. 2012, 430, 1–17.
- Kubas, H.; Schäfer, M.; Bauder-Wüst, U.; Eder, M.; Oltmanns, D.; Haberkorn, U.; Mier, W.; Eisenhut, M. Multivalent cyclic RGD ligands: Influence of linker lengths on receptor binding. Nucl. Med. Biol. 2010, 37, 885–891.
- Maresca, K.P.; Hillier, S.M.; Femia, F.J.; Keith, D.; Barone, C.; Joyal, J.L.; Zimmerman, C.N.; Kozikowski, A.P.; Barrett, J.A.; Eckelman, W.C.; et al. A Series of Halogenated Heterodimeric Inhibitors of Prostate Specific Membrane Antigen (PSMA) as Radiolabeled Probes for Targeting Prostate Cancer. J. Med. Chem. 2009, 52, 347–357.
- Liolios, C.; Buchmuller, B.; Bauder-Wüst, U.; Schäfer, M.; Leotta, K.; Haberkorn, U.; Eder, M.; Kopka, K.; Buchmuler, B.; Schaefer, M. Monomeric and Dimeric 68Ga-Labeled Bombesin Analogues for Positron Emission Tomography (PET) Imaging of Tumors Expressing Gastrin-Releasing Peptide Receptors (GRPrs). J. Med. Chem. 2018, 61, 2062–2074.
- Liolios, C.; Shegani, A.; Roupa, I.; Kiritsis, C.; Makarem, A.; Paravatou-Petsotas, M.; Pelecanou, M.; Bouziotis, P.; Papadopoulos, M.; Kopka, K.; et al. Synthesis, characterization and evaluation of 68Ga labelled monomeric and dimeric quinazoline derivatives of the HBED-CC chelator targeting the epidermal growth factor receptor. Bioorganic Chem. 2020, 100, 103855.
- Wu, Z.; Li, Z.-B.; Cai, W.; He, L.; Chin, F.T.; Li, F.; Chen, X. 18F-labeled mini-PEG spacered RGD dimer (18F-FPRGD2): Synthesis and microPET imaging of αvβ3 integrin expression. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 1823–1831.
- Fersing, C.; Bouhlel, A.; Cantelli, C.; Garrigue, P.; Lisowski, V.; Guillet, B. A Comprehensive Review of Non-Covalent Radiofluorination Approaches Using Aluminum [18F]fluoride: Will [18F]AlF Replace 68Ga for Metal Chelate Labeling? Molecules 2019, 24, 2866.
- Liu, S. Radiolabeled Cyclic RGD Peptides as Integrin αvβ3-Targeted Radiotracers: Maximizing Binding Affinity via Bivalency. Bioconjugate Chem. 2009, 20, 2199–2213.
- Li, Z.-B.; Cai, W.; Cao, Q.; Chen, K.; Wu, Z.; Elhendy, A.; Chen, X. 64Cu-Labeled Tetrameric and Octameric RGD Peptides for Small-Animal PET of Tumor v 3 Integrin Expression. J. Nucl. Med. 2007, 48, 1162–1171.
- Hedhli, J.; Czerwiński, A.; Schuelke, M.; Płoska, A.; Sowinski, P.; La Hood, L.; Mamer, S.B.; Cole, J.A.; Czaplewska, P.; Banach, M.; et al. Synthesis, Chemical Characterization and Multiscale Biological Evaluation of a Dimeric-cRGD Peptide for Targeted Imaging of αVβ3 Integrin Activity. Sci. Rep. 2017, 7, 3185.
- Shi, J.; Kim, Y.-S.; Zhai, S.; Liu, Z.; Chen, X.; Liu, S. Improving Tumor Uptake and Pharmacokinetics of 64Cu-Labeled Cyclic RGD Peptide Dimers with Gly3 and PEG4 linkers. Bioconjugate Chem. 2009, 20, 750–759.