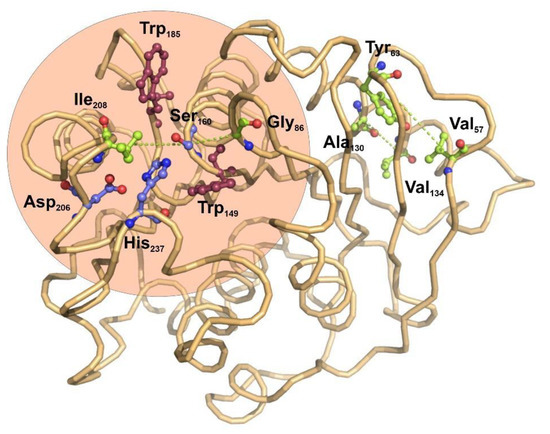
Far from the catalytic center, two other pairs of coevolving residues were determined: Tyr63-Val57 and Ala130-Val134. Both pairs of hydrophobic residues are located in the interface between a beta-strand and an alpha helix, opposite to the active center.
4. Discussion
Plastic polyesters are widely used polymers for the manufacturing of daily objects and their accumulation in the environment is a relevant problem. The currently implemented valorization measures are clearly insufficient to give a timely answer to plastic pollution in earth and aquatic environments. In this context, the importance of plastic bioremediation processes by microorganisms or microbial enzymes has been increased in the last decade [
48]. The first evidence for the microbial degradation of polyester plastics such as PET was described in
Thermobifida fusca, a thermophilic bacterium isolated from decaying organic matter [
49]. The enzyme responsible for this degrading activity was characterized as a serine hydrolase, functionally related to triacylglycerol lipases [
50]. In the following years, several microorganisms and enzymes able to degrade PET and other polyesters were described [
42,
47,
51]. The most well studied system is the PETase isolated from the marine bacterium
I. sakaiensis, which showed an increased degradation yield when compared with other similar enzymes [
20]. However, the number and diversity of polyester-degrading enzymes is expected to be expanded in the following years with the onset of the high-throughput metagenomic analysis of natural samples [
21,
52].
PETases and other plastic-degrading enzymes belong to the serine-hydrolase family, a widely distributed group of catalysts characterized by their relatively low substrate specificity and plethora or different activities [
11,
13]. Despite this catalytic diversity, their molecular structure is strongly conserved and, in many cases, shows no clear features that could classify a particular protein into a specific catalytic group. In this work, we performed a structure-based analysis of polyester hydrolases using the PETase from
I. sakaiensis as a reference, with the objective of finding structural features that could be related with the protein function. The generated knowledge could be applied, either for the screening and discovery of new members of the protein family or for the engineering and improvement of already known enzymes.
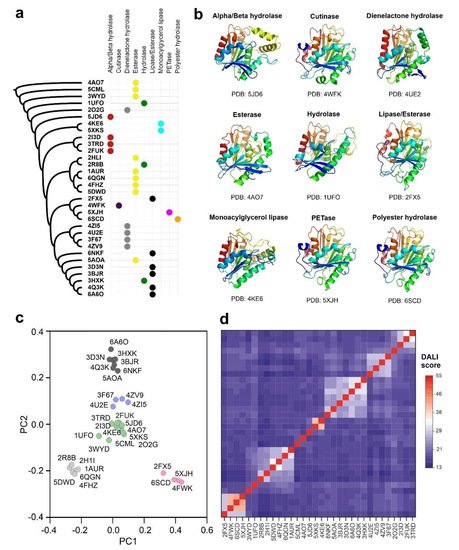
Our data generated by structure-based search, concluded that the alpha/beta fold observed in PETase is distributed across different proteomes. However, this particular folding is more associated with hydrolytic enzymes that act over branched polyesters in the form of high-molecular weight polymers. This group includes the bacterial cutinases and lipases, together with the already characterized polyester-hydrolases [
53]. Among them, the lipase from
P. mendocina has a particular interest, since it has been already characterized as active over polymeric complexes such as lignin for enzymatic bleaching [
43]. This bacterial alkaline lipase has an unusual structure, without the characteristic lid domain that covers the catalytic center [
23]. Other interesting structural homologs are represented by the family of cutinases, where the thermostable calcium-dependent cutinase from
S. viridis is the most relevant example. Despite its non-canonical mechanism and structure, the PET hydrolytic activity of this enzyme has been demonstrated over a wide range of low-density PET polymers [
16,
42]. Comparing the structural homologs and their enzymatic activity we can conclude that polyester hydrolases active over synthetic PET and other polymers would require the presence of an open active center with direct contact to the solvent [
10]. These results justify the low PET degradation efficiencies observed in canonical lipases where the active center is not directly accessible to the solvent. However, recent evidences showed that this catalytic limitation for lipases in PET degradation could be circumvented by synergistic associations with other enzymes, exemplified by a natural consortium formed by five species of
Bacillus and
Pseudomonas which is able to growth on PET as a sole source of carbon [
54].
PET-degrading enzymes are the result of a divergent evolution of hydrolases that, due to their relative substrate promiscuity, have been specialized to degrade very different ester polymers [
12]. At this point, it is difficult to state if the existence of specific PET-degrading enzymes, as the one isolated from
I. sakaiensis, is a result of environmental pressure or just an adaptation to a wide range of substrates. To infer the importance of amino acid coevolution in this group of enzymes, we performed an analysis with a selected group of PETase structural homologs. The problems derived from covariance models when protein sequences are analyzed, were minimized by the application of the visualCMAT algorithm that detectcs coevolution pairs in structure-based sequence alignments [
28]. Our results showed that with the exception of the catalytic Serine residue, the remaining coevolution pairs among the analyzed structures were amino acids located within structural elements, and located at distances not compatible with a molecular interaction. This fact is in agreement with the previous observations obtained in a large cohort of protein domains extracted from the PFAM database [
55]. Using the
I. sakaiensis PETase as representative structure, two coevolving residue pairs Ser160-Gly86 and Ser160-Ile208 were located in the boundaries of the catalytic pocket. Both coevolutionary pairs constitute a network which is spatially close and statistically coupled, suggesting an important role in the structure and function of the esterase center [
56]. However, no coevolution signal was detected between the residues involved in the catalytic triad, Ser160, Asp206 and His237. Since this triad is directly involved in the protein function and any variance could modify the catalytic properties of the enzyme, this constitutes a constraint that does not allow any variation and thus does not allow any covariation [
57]. Interestingly the remaining coevolving pairs Val57-Tyr63 and Ala130-Val134 are located in a region opposite to the catalytic center and involved in the structural alpha-beta packing of the enzyme. These coevolution pattern suggest a structural constraint in the alpha/beta fold that stabilizes the protein core without modifying the catalytic center, as already has been observed in other proteins such as synthetases or transcription factors [
57,
58].
However, it is still not clear what are the structural features that make PETase from
I. sakaiensis so efficient and unique in the PET degradation when compared with other similar enzymes with polyester hydrolase activity. These features are not related to the amino acids involved in enzyme activity or with the protein folding, since they are well conserved in other esterases [
11,
13]. We postulated regarding the involvement of the surface electrostatics in the interaction with polymeric substrates and in the overall catalytic yield. The comparison of the electrostatic potential function at the surface of the selected group of carboxyl esterases showed that PETase has a unique profile, measured as the electrostatic distance with other enzymes. This profile is partially shared with the bacterial cutinase from
S. viridis and the lipase-like enzyme from
P. mendocina. Combining the electrostatic information with the docking experiments, we clearly connected the presence of a positively-charged hinge in the surface of PETase structure (Asn233, His237, Asn 244, Asn246 and Arg280 residues) with the putative substrate-binding site ( and ). Based on an analysis of the results, we proposed that the increased efficiency of
I. sakaiensis in the degradation of PET and other polymers is related with the enhanced affinity of the substrate for the protein surface. The substrate-enzyme interaction is mediated by the establishment of a stretch of cation-pi interactions between the aromatic rings of phthalate groups and the positive charged hinge along the protein surface. The positively charged surface hinge is absent in less efficient enzymes, such as the polyester hydrolase from
P. aestusnigri, and only partially present in the
P. mendocina lipase and
S. viridis cutinase. In the
P. aestusnigri polyester hydrolase, experimental evidence showed the inefficiency of the enzyme to depolymerize high-density PET, and the limited activity over the amorphous polymer [
18]. The absence of the positively charged hinge in this enzyme suggests a limited substrate interaction when compared with
I. sakaiensis PETase, which is more evident in the case of crystalline PET.
Since the discovery of PET hydrolases, protein engineering has been applied to the improvement of their catalytic properties. Structural biology data combined with computer methods for protein design have been mainly focused in the improvement of protein stability. Son and coworkers applied a rational protein engineering method based on sequence homology to design a
I. sakaiensis PETase mutant (S121E-D186H-S242T-N246D) that showed prolonged enzymatic activity over 20 days [
59]. Moreover, Cui and coworkers developed a computational strategy for the mutational analysis of PETase and the design of a multiple mutant with enhanced thermostability when compared with the wild-type protein. This mutant designated as “duraPETase” contains the substitutions S214H-I168R-W159HS188Q-R280A-A180I-G165A-Q119Y-L117F-T140D, and it was able to degrade amorphous PET, but also longer polymers such as PBT with an optimum reaction temperature of 40 °C [
60]. However, both engineered PETase mutants showed low efficiency in the degradation of crystalline high-density polymers, suggesting that the compactness of the material limits the protein access to the free chemical groups. To solve this problem, some authors have proposed the use of enzyme cocktails. The rate of PETase hydrolysis could be significantly increased in the presence of proteins such as hydrophobin RolA [
61]. Hydrophobins represent a class of small fungal protein that has a high surface-active substance and can spontaneously self-assemble at hydrophilic-hydrophobic interfaces, increasing the exposure interface [
61].
Our results suggest that the interaction between the enzyme and the substrate could be improved by rational design of the binding interface, since the polyester hydrolases showing a higher PET degradation efficiency contain an interaction surface hinge with a positive charge (). This strategy could be useful for the degradation of high-density polyesters harboring aromatic residues, but also can open a new avenue for de novo design of more efficient depolymerases, based on the structural chassis of bacterial PETases and lipases.