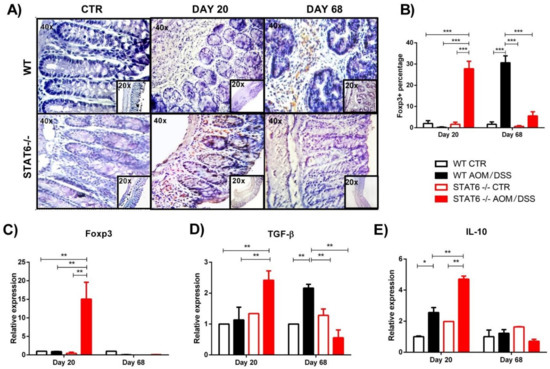
Next, we analyzed the colonic mRNA expression of Foxp3. Similar to the immunohistochemistry results, the relative expression of Foxp3 was significantly higher in the STAT6−/− AOM/DSS mice, compared to the WT AOM/DSS mice, at Day 20 (15 ± 6.4 vs. 0.9 ± 0.07, p < 0.05) (C). No differences were observed in the Foxp3 expression at Day 40 and Day 68 between the groups (C).
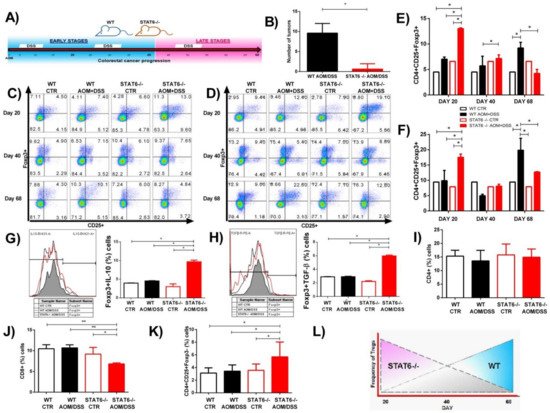
To corroborate our findings in the peripheral blood and spleen, we analyzed the gene expression in the colon of TGF-β and IL-10 cytokines that promote the suppressive function of Treg cells. We observed a significant increase level of TGF-β transcripts in the colons of the STAT6−/− AOM/DSS mice, compared to the WT AOM/DSS mice, at Day 20 (2.4 ± 0.5 vs. 1.13 ± 0.71; p < 0.001) (D). On the contrary, the expression of TGF-β mRNA was increased in the colons of the tumor-bearing WT mice, compared to the STAT6−/− AOM/DSS animals, at Day 68 of the CAC progression (2.1 ± 0.2 vs. 0.5 ± 0.4; p < 0.01) (D). On day 20, IL-10 levels were also increased in STAT6−/− AOM/DSS mice compared to the WT AOM/DSS mice (4.7 ± 0.2 vs. 2.5 ± 0.5, p < 0.01) (E). Interestingly, we observed similar IL-10 mRNA transcripts in the colon of both the WT and STAT6−/− AOM/DSS-treated mice during the advanced stages of tumor development (E). Taken together, our results demonstrate that STAT6 deficiency promotes the development of Treg cell responses during early CAC.
4. Discussion
STAT6 has important roles in the function and activation of immune cells. Here, we showed that STAT6 deficiency is necessary for controlling tumor growth in a model of CAC. Mechanistically, the absence of STAT6 resulted in an increased accumulation of local and peripheral Treg cells and overexpression of molecules associated with the function of Treg cells during the initial stages of CAC. These data are in accordance with our previous analyses, where the colons of STAT6
−/− mice showed a reduction in cell infiltration and decreased production of proinflammatory markers and cytokines in the initial tumor stage [
13]. Early depletion of Treg cells during CAC development in STAT6
−/− mice restores tumor growth, along with inflammatory infiltration in the colon. These data identify STAT6 as a critical pathway for the induction and function of Treg cells during CAC progression.
IL-4 and IL-13 are canonical inducers of STAT6 activation when dimers of STAT6 become phosphorylated and are translocated to the nucleus, where they activate or repress target genes. Phospho-STAT6 (p-STAT6) levels have been commonly detected in the colon of patients with clinically detectable CD or UC, and tumoral p-STAT6 is positively correlated to a clinical-stage and poor prognosis of human CRC [
9,
12]. The STAT6 signaling pathway favors the expression of anti-apoptotic proteins [
11] and promotes the proliferation of polyp epithelial cells in the colon [
12]. Similarly, the persistent activation of STAT6 modifies the expression of proteins involved in epithelial barrier permeability and interrupts tight junction integrity, resulting in the recurrent exposure of luminal microbiota, favoring inflammation and CAC development [
12,
28]. While these findings suggest the intrinsic importance of STAT6 in regulating tumor growth, our studies have found an important role of STAT6 in non-tumor cells for controlling tumor growth. AOM/DSS administration resulted in a significant decrease in inflammation and tumor development under STAT6 deficiency. In addition, in the STAT6
−/− animals, we found a remarkable increase in the frequency of Treg cells during early CAC, relative to the WT mice. The relationship between STAT6 and immune cells was shown to be important in a model of oxazolone-induced colitis, where T cells, macrophages, and natural killer T cells exhibit increased STAT6 phosphorylation during colitis development [
28]. Additionally, ApcMin/+Stat6
−/− mice developed few polyps, with a reduced proliferation at the small intestine and MDSCs expansion, decreased PD-1 expression in CD4+ cells, and strong CD8-mediated cytotoxic response [
29].
Treg cells have an oncogenic role in tumor progression through the suppression of antitumor immunity and prevention of an active cytotoxic process [
30]. Chemokines and cytokines are released in the tumor microenvironment (TME) by cancer cells and tumor stroma-infiltrating cells, leading to the recruitment of Treg cells. In the TME of many tumors, Treg cells suppress effector immune responses, overwhelming the anti-tumor activity mediated by natural killer cells and cytotoxic CD8+ T cells. However, the role of Treg cells in CRC is controversial. Some reports relate Foxp3+ cell infiltration with poor clinical outcomes [
31,
32,
33]. The colonic increase in Foxp3(+) cells is significantly higher in patients with CRC, compared to healthy controls or patients with inflammatory bowel disease [
34], and Treg cells with a higher expression of several molecules that correlate with suppression, such as Tim-3, LAG-3, TGF-β, IL-10, CD25, and CTLA-4, are observed in CRC patients [
32]. In contrast, a local accumulation and Foxp3 (+) cell density is associated with an improved survival rate and is considered as a good independent prognostic biomarker in the initial stage of colorectal cancers [
19,
35,
36]. The role of Treg cells in CRC seems to be dependent on the co-existence in the tumor tissue and the time of action of different subsets of Foxp3-expressing cells. A study identified two types of Treg cells in CRC, Foxp3
hi Treg cells and Foxp3
lo non-suppressive T cells [
20]. The latter are characterized by secreted inflammatory cytokines along with the instability of Foxp3 and the absence of CD45RA expression, a naive T cell marker [
20]. In the present study, we found that in early CAC development, Treg cells were efficiently recruited in STAT6
−/− colons, whereas increasing Foxp3 (+) cells in the colon of WT mice were detected only in the late stages of CAC. Interestingly, our previous results for WT mice showed that Treg cells from the late stages of CAC displayed an activated phenotype by expressing PD1, CD127, and Tim-3, along with an increased suppressive capacity in T-CD4+ and T-CD8+ cells. In contrast, Treg cells from WT mice in early CAC were scant and less suppressive [
21]. Due to the lack of STAT6
−/− Foxp3EGFP reporter mice, we were not able to determine the suppressive capacity of Treg cells in vitro that developed under STAT6 deficiency. However, the immunohistological analyses and H&E staining showed that the STAT6
−/− CAC-induced mice displayed decreased inflammatory infiltrate, with less destruction of the intestinal muscle and mucosa, supporting the idea that immunoregulatory mechanisms are taking place. Furthermore, the significant decrease in CD8+ T cells and the positive correlation between the frequency of Treg cells and the transcription levels of Foxp3, TGF-beta, and IL-10 in STAT6
−/− colons indicate that the latter is a consequence of the modulating function of Treg cells during CAC progression.
Some authors have suggested that distinct subpopulations of tumor-infiltrating Foxp3 (+) T cells contribute in opposing ways to the determination of CRC prognosis [
37,
38]. During the early CAC development, epithelial tight junction dysfunction promotes enhanced gut permeability, resulting in the deregulation of the interactions between the intestinal epithelial cells, immune cells, and gut microbiota. Continual exposure to luminal microbiota leads to intestinal inflammation, characterized by the recruitment of innate immune cells, the release of inflammatory mediators, and the subsequent generation and expansion of T-helper 17 (Th17) cells [
39]. The local increase of Treg cells in early CAC could suppress or prevent tumor formation through Th17 cell suppression. Here, we use an anti-CD25 monoclonal antibody (PC61) to deplete Treg cells only during early CAC. Interestingly, we observed histological damage and tumor growth recovery during STAT6 deficiency, supporting the idea that Treg cells developed in STAT6
−/− mice are protective against colon tumorigenesis. This finding seems to be compatible with that of a recent study showing that the administration of the natural compound, Parthenolide, during experimental colitis significantly relieved colon inflammation and improved the colitis symptoms [
40]. The protective effect of this molecule was associated with an increased frequency of colonic Treg cells, a downregulation of the ratio of colonic Th17 cells, together with a more abundant gut microbial diversity and flora composition [
40]. One possible reason for the protection observed in the STAT6
−/− mice during CAC progression could be the alterations in gut microbiota. Thus, this condition needs further research. Because the balance between Th17/Treg cells and these regulatory factors is decisive in CAC progression, it could be interesting to determine if the tumor growth observed in STAT6
−/− mice after Treg depletion is Th17-mediated.
Depending on the grade of alteration during CRC progression, Treg cells may modify their phenotype and exert different effects on cancer progression. Foxp3 (+) cells infiltrating colorectal carcinomas could be associated with their capacity to suppress tumor-promoting inflammatory immune response caused by infectious stimuli from bacterial translocation through the mucosal barrier [
41]. In a number of murine models, adoptively transferred Treg cells prevent the onset of colitis or treat established colitis [
42,
43,
44], and the in vitro expansion of Treg cells from the blood of patients with CD is considered as a feasible adoptive cell therapy for this disease [
45,
46]. The significantly higher percentages of Treg cells found in STAT6
−/− CAC-induced colons, compared with those found in WT colons, suggest that STAT6 limits Treg generation and recruitment by undetermined mechanisms. Additionally, a previous study from our laboratory demonstrated that the number of circulating CD11b+Ly6C
hiCCR2+ monocytes and CD11b+Ly6C
lowLy6G+ granulocytes was decreased in a STAT6-dependent manner [
13]. Additionally, a significant reduction in the expression of the chemokines, CCL9, and CCL25, and the chemokine receptor, CXCR2, both involved in the recruitment of inflammatory cells, was shown in STAT6
−/− mice during CAC progression. CCR2 is responsible for the recruitment of Ly6C
hi “inflammatory monocytes” to peripheral sites of inflammation, where they display inflammatory, phagocytic, and proteolytic functions [
47]. Thus, the STAT6 pathway in Treg cells has an intricate outcome and should be addressed in the context of an inflammatory environment. However, the local increase of Foxp3 (+) cells could be used to suppress antitumor immunity in the final phase of tumor formation. In our lab, when the Treg cells were depleted during the second DSS cycle of experimental CAC in WT mice, a reduction of 50% of Treg cells resulted in a better prognostic value, with a significant reduction in the tumor load [
22]. In contrast, in the present study, an earlier Treg depletion slightly increased the tumor load in WT mice. All these results suggest that Treg cells have a dynamic behavior influenced by STAT6 during CAC development.
Recently, a study demonstrated that STAT6 plays a critical role in the generation of Treg cells induced by B cells (Treg-of-B cells) [
48]. STAT6 phosphorylation was associated with the capacity of Treg-of-B cells to alleviate inflammation in an animal model of asthma in vivo [
48]. In accordance with our results, in an allergic disease model, a high number of Treg cells in the lungs and spleens in STAT6
−/− mice, compared to WT animals, were associated with a decreased allergic response [
26]. However, it would be interesting to determine if the stability in the expression of Foxp3 and therefore the differentiation and functional properties of Treg cells are modified in a STAT6-dependent manner. Furthermore, the role of STAT6 in modulating different types of Treg cells (natural, induced, type 1 T regulatory cells, and Treg-of-B cells) may be useful to develop new therapeutic strategies for relieving CAC or CRC.
In conclusion, STAT6 seems to play a central role in the regulation of the activity of Treg cells, particularly during the initial stages of CAC development, through the modulation of intense inflammatory responses.