1. Connexin43-Formed Channels
The subunit of gap junction channels, connexin proteins, are widely expressed in the heart, as well as throughout the body [
1,
2,
3,
4]. Connexins are crucially important to cardiac electrophysiology, having direct and indirect assignments in facilitating the propagation of action potentials between cardiomyocytes [
5,
6]. Connexin 43 (Cx43 gene name
GJA1) is the main cardiac connexin, being largely expressed by cardiomyocytes, but also expressed by fibroblasts, myofibroblasts, and vascular cells in the heart [
7,
8,
9,
10]. The half-life of Cx43 is ~1.5 h, which is approximately 1000 times shorter than that of cardiac collagen—a prodigious rate of turnover hinting at the substantial and still not as yet fully understood functions of Cx43 [
6,
11,
12]. As is the case with all 21 expressed connexins that are encoded by the human genome [
13], Cx43 is a transmembrane protein, with four membrane-spanning domains and cytoplasmically located loop, amino- and carboxyl-terminal domains [
14].
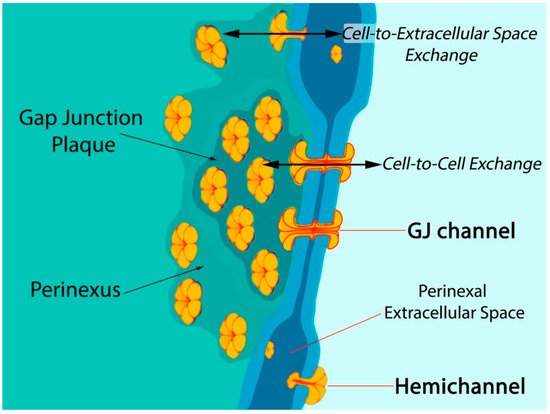
Six connexin subunits oligomerize during trafficking to the cell membrane to form a connexon channel, which when open is capable of transferring ions and other small molecules (typically < 1000 daltons) in a gated and relatively nonselective manner () [
5,
6]. Connexons formed by most connexins can occur in two states in the cell membrane, either as undocked hemichannels (HCs) or docked with another connexon from an adjacent cell to form a gap junction (GJ) channel. Consequently, connexons can perform two distinct information exchange functions: first, as undocked connexon HCs, which enable two-way exchange between the cell interior and the extracellular milieu; and second, as docked connexons in GJ channels providing a regulated pathway for cytoplasmic exchange between cells without recourse to the extracellular space. Undocked HCs in the cell membrane comprised of Cx43 are typically closed but can open in response to various prompts including ischemic injury [
15,
16,
17,
18,
19,
20,
21,
22].
Figure 1. Model of gap junction between two cells. Connexon hemichannels dock to form intercellular channels coupling the cells, enabling cytoplasmic exchange of ions and small molecules typically less than 1000 Da in molecular weight. Undocked hemichannels, concentrated in the perinexus surrounding the gap junction [
23], are typically closed but can open in response to prompts like ischemic stress. Open hemichannels thereby underpin flow of channel-permeant molecules from the cytoplasm into the extracellular space.
The biology of connexin structure and function and particularly that of Cx43 and its functions in intercellular communication have been covered extensively by us and others [
6,
7,
9,
10,
24,
25]. A focus of this review is the biology and pathophysiology of HCs formed by Cx43 in the myocardium, as well as the growing literature on the opportunities and barriers to pharmacological targeting of these channels in the treatment of heart disease. There is a wealth of preclinical data indicating the potential for therapeutic benefit from targeting HC activity by drugs mimicking the sequence of Cx43 in experimental models of human pathology, including those involving injury to the skin, heart, and brain [
4,
25,
26,
27,
28,
29]. If translation of these Cx43-targeting drugs to the clinic is to occur, careful attention to addressing questions of how to safely and effectively deliver drugs based on short peptides is required.
2. Connexin43 and Myocardial Pathology
Numerous cardiac disease processes and pathologic mechanisms have been linked to Cx43 (recent reviews of this topic include [
9,
25,
29,
30,
31,
32]). Of these, the most studied and clinically significant are myocardial infarction (MI) and ischemia reperfusion (I/R) injuries of the heart linked to this syndrome [
6]. In the post-MI environment, Cx43 expression is reduced and localization of Cx43 is disturbed (lateralized) in myocytes at the infarct border from as soon as 1 h following ischemic insult [
33]. Long-term Cx43 downregulation and lateralization are thought to contribute to the increased potential for electrical conduction disturbance in hearts subject to ischemic injury, particularly re-entrant arrhythmias triggered in tissues surrounding a MI—the infarct border zone (IBZ) [
34,
35].
Among the post-translational changes that appear to be significant to the injury status of the heart are alterations in the phospho-status of Cx43. Ek-Vitorin and colleagues found that Cx43 phosphorylated at a consensus PKC (protein kinase C) site, serine 368, was retained at intercalated disks during early ischemia and that this retention was associated with cardioprotection [
36]. Pertinently, phosphorylation of Cx43 at S368 (Cx43 pS368) is correlated with reduced activity of Cx43-formed HCs [
36,
37,
38]. Phosphorylation of Cx43 associated with mitochondrial membranes has also been linked to I/R injury [
39]. Phosphorylation at S368 and certain other serines in CT (e.g., Serines 262 and 373) are dependent upon dephosphorylation of a serine 365, “gatekeeper” amino acid [
40]. The downstream changes to Cx43 that ensue following S365 dephosphorylation result in changes to the gating and perm-selectivity of Cx43-formed channels, as well as the dysregulation of Cx43-ZO-1 interactions [
41]. Another post-translational modification significantly affecting Cx43 is ubiquitination [
42]. One of first pieces of evidence that Cx43 is ubiquitinated was provided by Laing and Beyer [
43]. Cx43 ubiquination acts as a signal for gap junction endocytosis by recruiting the ubiquitin binding protein Eps15 (epidermal growth factor receptor substrate-15) [
44]. Significantly, Cx43 ubiquitination has been proposed to control its postendocytic sorting from early endosomes to lysosomes, controlling degradation of Cx43 [
45]. Ubiquitinated forms of Cx43 are recognized by the endosomal sorting complex required for transport (ESCRT) sorting system, which is responsible for sorting into endosomes and exosomes [
45].
Post-translational modifications to Cx43 appear to be of particular significance at the edge of GJs—in the perinexus—a specialized nanodomain where Cx43 HCs are concentrated () [
6,
23,
46]. During the acute phase of an MI, large-scale opening of Cx43 HCs occurs [
18,
47]. HC opening may be also exacerbated during the reperfusion phase of an I/R injury. Pro-inflammatory and injury-spread signals resulting from unregulated opening of Cx43 HCs are likely crucial to the generation and severity of I/R damage [
6,
10,
48,
49]. The flux of cytoplasmic contents released by HCs is thought to contribute to a “bystander effect”, wherein cell loss and necrotic damage are caused in otherwise healthy tissue adjacent to the primary site of injury. Intercellular coupling by GJs may also contribute to injury spread via the bystander effect [
50,
51,
52]. Examples of tissues susceptible to the “bystander effect” include myocardium-at-risk [
53], cardiac muscle tissue surrounding the ischemic core of an MI. Adenosine triphosphate (ATP) is thought to be one of the more consequential pro-inflammatory molecules released by HCs [
17,
54,
55,
56]. Extracellular gradients of ATP facilitate directed migration by neutrophils to the sites of injury, disease, or infection [
57]. This purinergic signal also induces production of extracellular nets—a lethal behavior elicited by inflammatory cells that likely contributes to myocyte death and the extent of myocardium-at-risk following MI [
58].
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a pathology that is likely directly impacted by Cx43 expression and phosphorylation; ARVC patients experience a loss of desmosomes, a specialized cell–cell junction [
59,
60,
61]. Among key proteins in desmosomes is desmoplakin—the gene-encoding desmoplakin was the first desmosomal gene to be linked to ARVC [
62]. When this protein is deleted from cardiomyocytes, the cells exhibit large reductions in GJs and Cx43, as well as changes to Cx43 phospho-status, associated with loss of intercellular communication [
63]. A mutation of another desmosomal protein, plakophilin-2 (PKP2) has shed further light on ARVC disease mechanisms [
61]. Cardiomyocyte-specific knockout of PKP2 in mice results in generation of an arrhythmogenic substrate (Ca
2+ dysregulation) that was apparent at time points before overt structural changes in myocardium associated with ARVC-like disease (e.g., fibrosis). Interestingly, Cx43 ablation relieved these functional deficits in the ventricle. Moreover, treatment with a selective peptide-based blocker of Cx43 HCs normalized arrhythmogenic Ca
2+ dynamics—suggestive of a role for HCs in the formative stages of ARVC pathogenesis.
Duchenne muscular dystrophy (DMD) is an X-linked recessive disorder resulting from loss-of-function of dystrophin [
64]. In skeletal and cardiac myocytes cells, dystrophin is a membrane-associated protein that links the cytoskeleton and the surrounding extracellular matrix, thereby providing mechanical integrity during muscle cell work [
65]. The absence of dystrophin triggers a cascade of biochemical changes that ultimately leads to the death of muscle cells and fibrosis associated with myofibroblast proliferation. The primary culprit in muscle loss appears to be intracellular calcium overload and the triggering of oxidative stress pathways leading to cell death. This chain of events results in over 50% of DMD patients experiencing cardiomyopathy by the age of 10, with over 90% of patients experiencing cardiac dysfunction by the age of 18 [
66]. The particular danger in DMD in relation to Cx43 lies in progression to dystrophic cardiomyopathy, a pathology strongly affecting Cx43 expression and function [
67]. Dystrophic cardiomyopathy patients experience Cx43 remodeling away from intercalated discs to lateralized positions, increased Cx43 expression, and a propensity to develop disturbances to cardiac conduction. Recent evidence suggests that unregulated opening of HCs is central to the role of Cx43 in the pathogenesis of DMD [
68,
69]. S-nitrosylation and reduced phosphorylation of Cx43 at serines S325/S328/S330 have been reported to be associated with HC activation and arrhythmogenesis and ventricular remodeling in the Mdx mouse model of DMD [
68]. A recent study has suggested that inhibition of aberrant HC activity in skeletal muscle macrophages neighboring Cx43 nonexpressing fibers in symptomatic DMD mice leads to prevention of Cx43 remodeling in the heart and protection from the loss of skeletal and cardiac muscle cells that characterize this disease [
70].
Yet another example of probable involvement of Cx43 HCs in cardiac disease processes comes from a study of myocytes expressing altered nuclear lamin A/C proteins, mutations associated with laminopathy—a disease in humans leading to heart failure and arrhythmias [
71]. Cultured neonatal rat cardiomyocytes expressing pathologic human lamin A/C mutations exhibit altered microtubule structure, hemichannel localization and beating force, frequency, and contractile amplitude. The common endpoints of diseases such as ARVC, DMD, and laminopathy are muscle cell death and irreversible replacement of lost myocardium with scar tissue. The mounting evidence suggests that HC activation is a precursor to this fibrotic replacement, often in association with oxidative stress, dysregulated intracellular Ca
2+ dynamics, disturbances to cardiac conduction, and arrhythmias.