MAG is a transmembrane glycoprotein produced by oligodendrocytes in the CNS [
61]. Some discrepancies exist in the function of MAG in response to injury and disease. Genetic deletion of MAG reduced corticospinal tract (CST) axon sprouting, an important tract widely studied in SCI models [
62]. Conversely, MAG has been extensively used as an inhibitory substrate for axon elongation in mature neurons [
63].
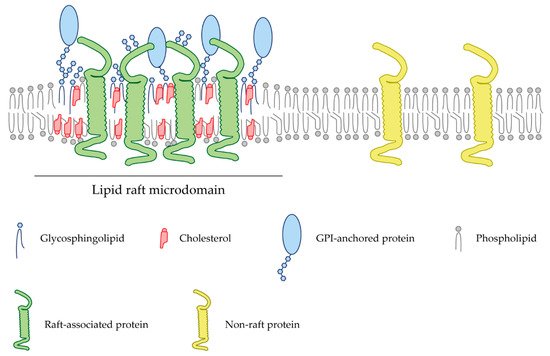
Gangliosides are glycosphingolipids that carry one or more sialic acid residue(s). A particularity of brain gangliosides is that due to the biophysical properties of their ceramide moiety, they tend to aggregate in lipid microdomains [
64]. Among the gangliosides expressed in the brain, GD1a and GT1b have been shown to interact with MAG to induce the inhibition of axon regeneration [
65]. Both gangliosides reside in membrane rafts, where they interact with the GPI-linked NgR2 protein to inhibit axon outgrowth after CNS damage [
66]. Isolated MAG from whole-brain extracts and oligodendrocytes are found in lipid raft compartments [
67]. Localization within these domains is dependent on cell membrane cholesterol, as demonstrated after MβCD treatment [
67]. Moreover, the localization of NgR1 and GT1b within cholesterol-enriched membrane microstructures is required for the interaction of MAG and these receptors [
67]. This molecular environment may facilitate the downstream signaling required for the response to MAG. Furthermore, MAG or the antibody against GT1b and GD1a promote the recruitment of p75NTR to lipid rafts, suggesting the importance of these gangliosides in the recruitment of p75NTR to cholesterol-enriched microdomains [
68]. Disruption of lipid rafts abolishes the inhibitory effects induced by MAG and Nogo peptides in postnatal cerebellar neurons, indicating that raft microdomains play an important role in the initiation of its signal transduction during neurite outgrowth [
68]. MAG also binds to the lipoprotein-receptor-related protein-1 (LRP1), which is involved in the activation of RhoA, thereby facilitating the collapse of growth cones and inhibiting neurite outgrowth [
69]. MAG-mediated activation of RhoA may involve both LRP1 and p75NTR, as its binding induces colocalization of both receptors [
69]. In neurons and neuronal-derived cell lines like PC12 and N2a cells, LRP1 has been found to partially localize in lipid rafts [
70]. Upon raft disruption with MβCD, LRP1-mediated signaling is impaired, while its ligand-binding capacity remains intact [
70]. This indicates that MAG plays an important role in recruiting LRP1 and p75NTR receptors to lipid rafts, where the MAG-dependent signaling cascade that promotes axon retraction is initiated. Therefore, targeting lipid raft stability can become a novel strategy to prevent axonal retraction after damage induced by MAG and their receptors.
Many pieces of evidences suggest that integrins participate in the regulation of neurite extension, axonal guidance and neuronal migration [
71]. β1 integrin has been shown to act as a receptor for MAG, mediating growth cone responses independent of NgRs in rat hippocampal neurons. β1 integrin signaling has been demonstrated to be required for both MAG-induced attraction in developmental stages of the CNS and repulsion in mature CNS [
72].