The important role of human gut microbiota in liver diseases has long been recognized as dysbiosis and the translocation of certain microbes from the gut to liver. With the development of high-throughput DNA sequencing, the complexity and integrity of the gut microbiome in the whole spectrum of liver diseases is emerging. Specific patterns of gut microbiota have been identified in liver diseases with different causes, including alcoholic, non-alcoholic, and virus induced liver diseases, or even at different stages, ranging from steatohepatitis, fibrosis, cirrhosis, to hepatocellular carcinoma. At the same time, the mechanism of how microbiota contributes to liver diseases goes beyond the traditional function of the gut–liver axis which could lead to liver injury and inflammation.
- gut microbiota
- chronic liver diseases
- metabolites
- immune system
1. Background
Human gut microbiota contains trillions of microbes, contributing to human health and diseases through various routes of mechanism. Recent studies have discovered a myriad of novel functions of gut microbiota linking gut to liver [1][2][3]. With the advancement of high-throughput sequencing, including metagenomics, metatranscriptomics, metabolomics, proteomics, and culturomics, such ever-changing technologies usher the revelation of the tremendously diverse microbiome in humans, in both the intestines and extra-intestinal organs communicating with the gut (Figure 1). Human studies have demonstrated compositional alterations of microbiota in diseases versus health, including those intensively studied in inflammatory bowel disease (IBD), colon cancer, diabetes, and relatively less in liver diseases [4]. However, substantial efforts have been made to unravel the association versus causal relationship between microbiota and disease pathogenesis. In this review, we center on the recent advance in our perception of how gut microbiota contributes to chronic liver disease.
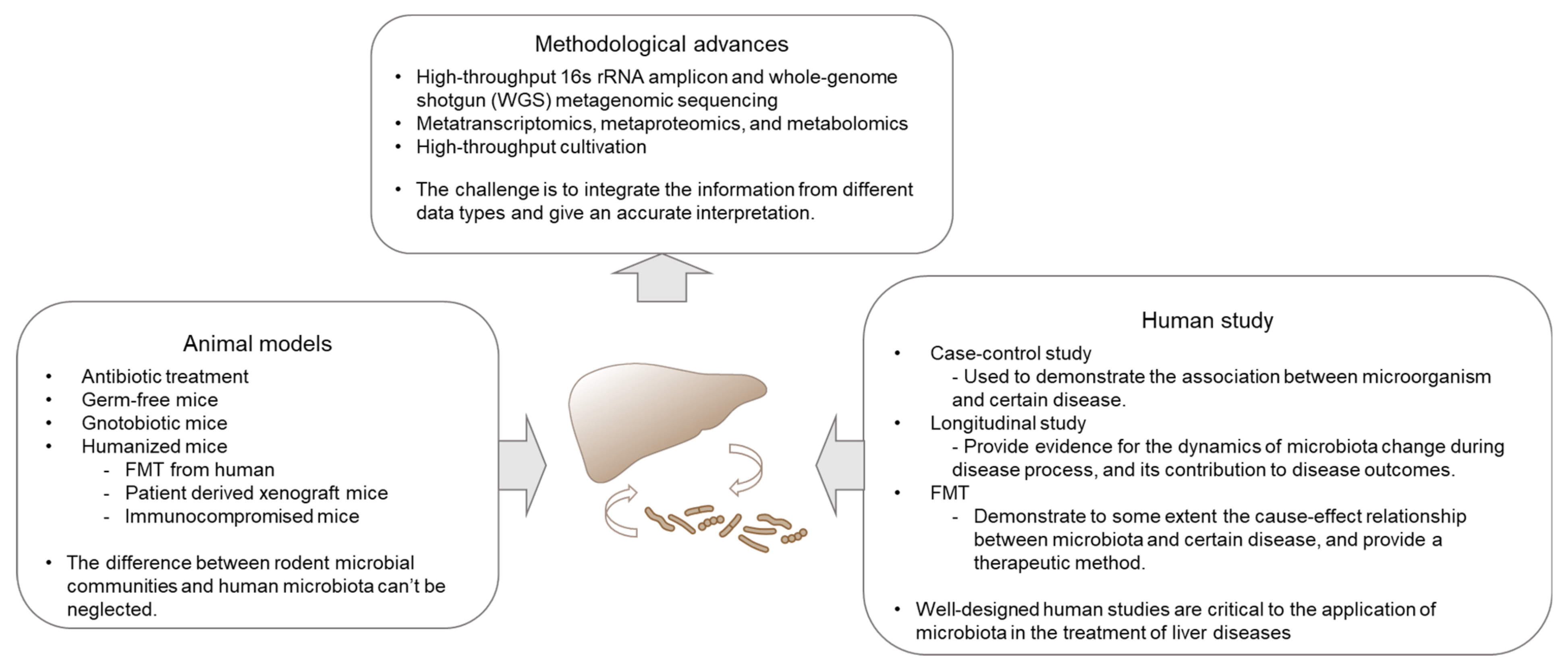
Figure 1. Technology development and the availability of a suite of tools facilitated perception and exploitation of gut microbiota in liver diseases. Multi-omics enables the exploration of the complexity and integrity of the gut microbiome in a spectrum of liver diseases. Mouse models serve as a conductive tool in mechanistic studies, especially the newly developed humanized mice, which resembles human in many aspects, including genetic, immunological and microbiome factors. Human studies reveal the association between microbiota and liver disease, which is the end-resort of microbiota study. Human trials are also critical in final testing of the preventive and therapeutic potential of microbiota in diseases.
Liver diseases cause ~2 million deaths per year worldwide [5], with cirrhosis and hepatocellular carcinoma as the leading cause of mortality and mobility. Chronic liver disease results from various etiologies, including virus infection, alcoholic liver disease (ALD), and non-alcoholic fatty liver disease (NAFLD). Chronic liver injury can cause liver fibrosis and cirrhosis, with high risk of hepatocellular carcinoma. Nearly 75% of the liver’s blood supply comes from the portal vein, which consists of blood from the intestines and spleen. This special physiological construction of the liver ensures its consistent interaction with the gut-resident microorganisms and their metabolites. Accumulating evidence has demonstrated a sophisticated link between the gut microbiota and the outcomes of chronic liver disease [6][7][8][9]. Gut microbiota has been shown to impact liver diseases through producing metabolic products and modulating host immune responses [10][11][12]. More recently, the alterations of gut microbiota in COVID-19 patients have been studied [13]. Acute liver injury has been reported to be common, around 70%, in patients who test positive for SARS-CoV-2 [14]. The possible contribution of gut microbiota in COVID-19 induced liver injury might be worth further analysis.
Remarkable advances in the understanding of microbiota and liver diseases have been made over the past decades. Building on cutting-edge technologies, we anticipate that our understanding of host-microbial interaction will continue to increase within the coming years. Meanwhile, the advancement in microbial-related diagnostics and therapeutics is envisioned for probing and treating liver diseases [15][16][17].
2. Clinical Evidence of Microbiota Alterations in Liver Diseases
2.1. NAFLD
NAFLD consists of a wide spectrum of liver diseases, ranging from steatosis to nonalcoholic steatohepatitis (NASH), liver fibrosis, and cirrhosis. The pathological process of NASH has been firstly proposed to be the result of “two hits”, which are represented by hepatic steatosis and lipid peroxidation [18]. This theory later evolved into “multiple hits”, comprising signals derived from the gut or the adipose tissue, such as endotoxin, adiponectin, IL-6, and TNFα [19]. Besides dietary and genetic factors, the gut microbiota has been demonstrated as an important novel factor in disease pathogenesis, involving in all aspects of the “multiple hits” [19][20].
The early disease modality leading to NASH is hepatic steatosis, which is viewed as the liver manifestation of metabolic syndrome. The gut microbiota has been linked to a variety of metabolic disorders, with disease-specific signatures [21][22][23][24][25]. Bacterial richness is inversely associated with metabolism-related phenotypes. Individuals with low bacterial richness were at higher odds for insulin resistance, dyslipidemia, and inflammatory phenotype [21]. A decrease in the proportion of Bacteroides was observed in obese subjects compared with lean subjects [26], although this finding is not consistent across all studies [27]. In addition, Bacteroides vulgatus was reported to impact the serum metabolome and associate with insulin resistance [28], and Bacteroides spp. was associated with hepatic steatosis [10]. These data suggest versatile roles of gut microbes in different diseases with similar hepatic pathologies.
More than a decade ago, systematic analysis of gut microbiota was conducted in fatty liver disease [20]. The relation between microbiota and NASH has been discovered in different population-based cohorts. In a pediatric cohort of NASH, Zhu et al. observed a significant increase of the Enterobacteriaceae family, accompanied with an increase in blood alcohol concentrations, compared to obese and healthy controls [29]. In view of that Enterobacteriaceae could produce ethanol toxic to the liver, this evidence proposed a microbiome-based mechanism to explain the histologic similarity between NASH and alcoholic hepatitis. By contrast, in another adolescent cohort of NAFLD and healthy controls, Mouzaki et al. found no statistical difference in Escherichia coli between two groups [30]. Besides, they found a decrease of fecal Bacteroidetes in NASH relative to simple steatosis and healthy controls, which is in agreement with the discoveries in obese patients [26]. Echoing these findings, an increased abundance of Bacteroidetes and reduced abundance of Firmicutes were associated with improvement in hepatic steatosis in a longitudinal study [31]. However, in a group of biopsy-proven NAFLD patients, Boursier et al. observed a higher abundance of Bacteroides in NASH and fibrosis patients, compared to those without NASH, suggesting a relation between Bacteroides and disease severity [7]. Recently, the high-alcohol-producing Klebsiella pneumoniae is found to be associated with NAFLD patients in a Chinese cohort [32]. Again, this data support that an alteration in the gut microbiome contribute to NAFLD through excessing endogenous alcohol production. Other studies, which found no significant difference at the phylum level, documented changes at the class or genus level [33][34]. The discrepancies across these studies might result from small numbers of study subject, intrinsic and extrinsic host factors, including age, gender BMI, environmental, geographical, and dietary factors, and even differences in methodology. Despite these variable findings, the correlation of microbiota and NASH has been well recognized.
Recently, the crosstalk between the microbiome and the human host was comprehensively investigated by integrating metagenome, metabolome and hepatic transcriptome and clinical features [10]. Microbial gene richness decrease was found to be an indicator of deleterious changes. A more complex model was proposed to explain the microbiome–host interplay, involving both metabolism and immune responses. For example, microbial metabolites such as phenylacetic acid (PAA), and microbial products such as Lipopolysaccharide (LPS) have been found to facilitate hepatic lipid accumulation and induce hepatocyte inflammation [10]. Further investigation revealed that Proteobacteria, Actinobacteria and Verrucomicrobia were positively associated with liver steatosis, whereas Firmicutes and Euryarchaeota were negatively associated with liver steatosis [10]. Overall, the microbial alterations in patients with liver diseases are summarized in Table 1.
Harnessing germ-free (GF) animal models, evidence is accumulating to show a causal effect of microbiota in the pathogenesis of metabolic diseases. Germ-free mice were protected against high fat diet (HFD) induced obesity, and conventionalization of GF mice leads to an increase in body-fat content and insulin resistance [35][36]. Later analysis found obese microbiome could increase energy harvest from the diet. This trait is transmissible through the colonization of germ-free mice with gut microbiota from obese mice (ob/ob mice) [37][38]. Moreover, gut microbiome composition can affect mouse response to HFD, manifested by changes in blood glucose levels and plasma concentrations of pro-inflammatory cytokines. Transplantation of microbiota from HFD mice who have hyperglycemia and higher levels of systemic inflammation can induce the same phenotype in recipient mice [39]. Fecal microbiome from patients with hepatic steatosis could also trigger steatosis in recipient mice [10], which further reinforces the significance of microbiota in the pathogenesis of NAFLD.
Table 1. Important studies of human microbiome and liver diseases.
| Study | Sample Types | Disease | Method | Major Findings |
|---|---|---|---|---|
| Mouzaki et al., 2013, Hepatology [30] | Stool | NAFLD | Quantitative real-time PCR | The first study assessing the microbiota in adult human NAFLD at different histological stages. Lower percentage of Bacteroidetes is associated with NASH. |
| Jerome et al., 2016, Hepatology [7] | Stool | NAFLD | 16s sequencing | Gut dysbiosis associates with the severity of NAFLD lesions. Bacteroides associated with NASH and Ruminococcus with fibrosis. |
| Loomba et al., 2017, Cell Metabolism [17] | Stool | NAFLD | Shotgun sequencing | A gut microbiome signature was identified to predict NAFLD with fibrosis. The abundance of Firmicutes and Proteobacteria was higher in mild/moderate fibrosis, and advanced fibrosis, separately. |
| Mutlu et al., 2012, Am J Physiol Gastrointest Liver Physiol [40] | Human sigmoid mucosa biopsy | ALD | Multitag-pyrosequencing | The first study showing the association between alcohol consumption and microbiome using non-culture method in human. Lower abundances of Bacteroidetes and higher ones of Proteobacteria was found in alcoholics. |
| Dubinkina et al., 2017, Microbiome [9] | Stool | ALD | Shotgun sequencing | Alcoholic dependence was inversely associated with Clostridiales. The expansion of Bifidobacterium and Lactobacillus was found in ALD patients, but the species were different between patients with and without cirrhosis. |
| Liu et al., 2004, Hepatology [16] | Stool | Cirrhosis | Quantitative bacteriological Culture | Early evidence of disturbed microecology in cirrhotic patients with MHE, with outgrowth of Escherichia coli and Staphylococcal species. |
| Chen et al., 2011, Hepatology [41] | Stool | Cirrhosis | 454 pyrosequencing 16s | The first major study of microbiome in cirrhosis. Fecal microbial diversity was lower in cirrhotic patients, with higher levels of Enterobacteriaceae and Streptococcaceae, and lower levels of Lachnospiraceae. |
| Bajaj et al. 2014, Journal of Hepatology [42] | Stool | Cirrhosis | Multi-tagged pyrosequencing | The first major study to compare the microbiome in compensated and decompensated cirrhosis. Introduction of CDR, which is associated with disease progression and prognostic. |
| Qin et al., 2014, Nature [43] | Stool | Cirrhosis | Metagenome sequencing | The major study characterized the gut microbiome in liver cirrhosis. Patient-enriched species are of buccal origin, with higher levels of Proteobacteria and Fusobacteria and fewer levels of Bacteroides. |
| Caussy C et al., 2019, Nat Commun. [44] | Stool | Cirrhosis (NAFLD-associated) |
16s sequencing | Bacterial signature was discovered to detect NAFLD-cirrhosis based on a prospective twin and family cohort. |
2.2. ALD
Alcohol has been well demonstrated to be a hepatotoxin, and alcoholic liver disease remains the most prevalent type of chronic liver disease worldwide [45]. The pathogenesis of ALD includes the toxicity of acetaldehyde, hepatic steatosis, and inflammation. Other factors, both genetic and non-genetic, contribute to interindividual variations in disease development. Among them, the gut microbiota plays a critical role in alcohol-induced liver damage. Alcohol abuse leads to bacterial overgrowth, dysbiosis, and gut barrier dysfunction. The subsequent translocation of bacterial products through portal vein stimulates inflammation and metabolic disorders in the liver [46].
Dysbiosis refers to the microbial imbalance in the gut. A pioneer study compared the types and numbers of bacteria in the aspirates from the jejunum of patients with and without alcohol abuse and revealed an increase in the number of microorganisms both anaerobic and aerobic, suggesting a role for bacterial overgrowth in the pathogenesis of chronic alcohol abuse [47]. Later, a study comparing the mucosa-associated colonic microbiome between alcoholics with or without ALD and healthy controls, observed gut microbiome dysbiosis in a subgroup of alcoholics, with lower abundance of Bacteroidetes and higher ones of Proteobacteria [40]. Altered fecal microbiome was also demonstrated in ALD-related cirrhosis compared to healthy subjects [41][48].
Murine models of alcoholic liver disease also showed an intestinal bacterial overgrowth and enteric dysbiosis in alcohol fed mice [49]. Moreover, compared with conventional mice, germ-free mice showed less liver injury after alcohol consumption [50], corroborating the critical role for gut microbiome in liver disease development. The causal effect of microbiota on ALD has been proposed based on the “leaky gut” theory. The intestinal barrier consists of a mucus layer, physical integrity, and immune defense. Increased intestinal permeability has been described in ALD patients [51]. Through a chromium-51 absorption test in non-intoxicated alcoholic patients, Bjarnason et al. discovered higher intestinal permeability than controls [52]. In alcoholics with chronic liver disease, through measurement of urinary excretion of lactulose and mannitol after oral administration, Keshavarzian et al. observed increased intestinal permeability compared to alcoholics with no liver disease and non alcoholics with liver disease [53]. This “leaky” gut phenotype was also supported by animal models of both acute and chronic ethanol administration [54]. Acute alcohol intake in rats could increase colonic permeability in 24 h and was associated with significant endotoxemia [55]. Ten weeks of alcohol gavage also induced gut leakiness in rats, resulting in endotoxemia and liver injury [56]. Evidence indicates that alcohol-induced gut leakiness and endotoxemia occurs prior to steatohepatitis, acting as a trigger for alcoholic steatohepatitis (ASH) [57].
Further analysis discovered a negative association between alcohol feeding and the expression of certain bactericidal protein in the host, which can be partially reversed by prebiotics treatment. Other manipulations, such as pectin treatment, and fecal microbiota transplantation (FMT), have been shown to be able to prevent alcohol-induced liver injury in mouse models [15]. A similar protective function of microbiota has been observed in ALD patients treated with probiotics and fecal transplantation, which leads to improved liver enzyme levels and better clinical outcomes [58][59]. Oral supplementation of Akkermansia muciniphila, which was found to be diminished by ethanol exposure both in mice and humans, can ameliorate experimental ALD and promote intestinal barrier integrity [60]. However, larger clinical studies across different populations with complete evaluation of benefits versus risks are needed before fecal transplantation or other therapeutic regimes built upon manipulation of the gut microbiota can be considered a routine clinical practice in the treatment of ALD.
2.3. Cirrhosis
Cirrhosis is the end stage of chronic liver disease, which is characterized by the formation of nodules of regenerative parenchyma, often accompanied by portosystemic shunts. Cirrhosis results from different etiologies, covering all the chronic liver diseases mentioned above, with significant differences between regions, genders, and socioeconomic status [5].
Changes in gut microbiota in patients with liver cirrhosis have been well documented in studies using various approaches. Early studies using bacterial culture found that the microecology was disturbed in cirrhotic patients with minimal hepatic encephalopathy (MHE), characterized by outgrowth of Escherichia coli and Staphylococcal species. This can be reversed by synbiotic treatment accompanied by a reduction in blood ammonia and amelioration of MHE [16]. A later study using 16S rRNA sequencing observed the distinct gut microbiome in liver cirrhosis, showing that at the phylum level, Bacteroidetes was reduced, while Proteobacteria and Fusobacteria were enriched in the fecal microbiota [41]. Among the bacteria enriched in cirrhosis, Enterobacteriaceae, Streptococcaceae, and Veillonellaceae were the most abundant families [41]. A more recent study using quantitative metagenomics revealed that the major change of gut microbiota derives from bloom of oral bacterial species within the gut [43]. Moreover, a combination of 15 biomarkers was used for discrimination of patients with liver cirrhosis from healthy subjects, highlighting the diagnostic potential of microbial markers for liver cirrhosis [43]. These microbial genes, with a high specificity to liver cirrhosis, have no overlap with the markers discovered in type 2 diabetes study [23], which suggests that such microbial features are disease specific.
Other studies have found a correlation between changes of microbiota and severity of disease. Bajaj et al. established a cirrhosis dysbiosis ratio (CDR) based on the ratio of autochthonous to pathogenic taxa, with a low CDR associated with worse disease state [42]. In a longitudinal analysis, comparing patients before and after hepatic encephalopathy (HE) development, CDR was also found to be decreased in patients after occurrence of HE [42]. Loomba et al. showed a shift in microbiome composition during disease progression from mild NAFLD to advanced fibrosis, with an increase in Proteobacteria and a decrease in Firmicutes at the phylum level. At the species level, E. rectale and B. vulgatus were the most abundant microorganisms in mild and advanced fibrosis, respectively [17]. They also identified 37 microbial species which distinguish different stages of the disease, suggesting the potential use of microbial markers as a tool in diagnosing and determining the stages of liver disease. Qin et al. found that the severity of liver cirrhosis correlated with the abundance of orally derived species in the gut [43]. In line with these results, studies trying to investigate the effect of gut bacteria on the gut-liver-brain axis by magnetic resonance imaging (MRI), found a positive correlation of Enterobacteriaceae and Streptococcacae with astrocytic changes. In addition, Porphyromonadaceae is associated with changes of neuronal integrity and edema [61].
Commonly used liver cirrhosis/fibrosis mouse models include chemical-based models, which is induced by carbon tetrachloride, thioacetamide or ethanol, diet-based models, mostly methionine-deficient and choline-deficient diet, and surgery-based models, as the common bile duct ligation [62]. Although differences exist between the cause of liver cirrhosis in human and in mouse models, significant change in the gut microbiome were observed in nearly each model [63]. The underlying mechanism of how microbiota effect liver cirrhosis has also been explored by using animal models, which include regulation of gut permeability to release bacterial products and regulation of liver metabolism [64]. However, with the large differences between intestinal microbiota in mice and humans [65], to translate the knowledge gained from mouse studies to human should be very careful [66].
2.4. Liver Cancer
The development of liver cancer is a multi-step process, involving factors including genetics, environmental factors, metabolism, and the immune system. Alterations in the microbiota have been reported to contribute to the development of cancer and modulate the efficacy of cancer therapy [67].
In a human study of cirrhotic patients with HCC, the fecal microbiota dysbiosis is characterized by an overgrowth of E. coli [68]. In human liver samples, helicobacter was detected in liver carcinoma, while no evidence of helicobacter can be found in patients without malignancy [69]. Studies in animal models have shown that a lack of gut bacteria prevented development of hepatocellular carcinoma (HCC) in different models [6][70]. In HCC mouse model using DEN (diethylnitrosamine) plus CCl4 treatment, LPS from the intestinal microbiota contributes to tumor growth by promoting liver cell proliferation, suggesting a role for gram-negative bacteria in tumorgenesis [6]. On the other hand, gram-positive bacterial strains were shown to be increased in mice fed with HFD, and deletion of gram-positive bacteria with vancomycin alleviated the development of HCC induced by DMBA (7,12-dimethylbenz(a) anthracene) and HFD [70].
In addition to the accumulating evidence suggestive of the relation between microbiota and cancer development, the potential role of microbiota in anticancer therapy has been studied [71][72]. In melanoma patients, higher gut microbiome diversity and a relative higher abundance of Ruminococcaceae are related with better response to anti-PD-1 immunotherapy [71]. Further study by transplantation (FMT) of stool from responders to anti-PD-1 therapy to germ-free mice showed reduced tumor growth and more importantly, improved responses to anti-PD-1 therapy [71]. In tumor-bearing mice, gut microbiota has been shown to shape the antitumor immune response through translocation into secondary lymphoid organs and thereby stimulating T helper cells [72]. However, no study directly tested the effect of modulating gut microbiota in the treatment of HCC. Targeting the gut–microbiota–liver axis represents an attractive therapeutic option for HCC treatment [73].
3. Microorganisms from Other Origins and Kingdoms beyond Fecal Bacteria
Besides fecal microbiota, microbiota from other origins have also been implicated in liver disease, including those from the saliva [74], colonic mucosa [75], sigmoid mucosa [76], and the small intestine [77]. Moreover, evidence suggests that other components of the gut microbiota, including fungi, archaea, and viruses, might play a role in the disease process [78]. Fungal dysbiosis has been illustrated in patients with liver cirrhosis [79]. After antibiotics treatment, fungal diversity was decreased, concomitant with higher prevalence of Candida [79]. A decrease of fungal diversity was also found in alcoholic patients. Overgrowth of Candida was discovered in different stages of alcoholic liver disease, including alcoholic hepatitis and cirrhosis patients [80]. Viruses represent another important collection of microorganisms residing in the gut of humans. Histological severity of NAFLD was associated with a decrease in gut virome diversity [81]. Although high inter-individual variations were found in gut virome composition, several Lactococcus phages were found to be decreased in patients with more severe NAFLD [81]. However, an increased fecal virome diversity accompanied by a decreased bacteria diversity was found in patients with alcohol-associated liver disease compared with controls [82]. Others reported only a modest link between fecal phages and liver cirrhosis characteristics [83]. The difference in the change of virome diversity across studies might result from the different types of liver diseases or from different analytical methods. In addition, the gut virome can influence host through interactions with bacteria. When interrogating the phage-bacterial correlations in patients with liver cirrhosis, a higher network complexity was found in controls in contrast to a lower one in cirrhotic patients [83]. Furthermore, a potential therapeutic effect of bacteriophages was shown in ALD [84]. The study showed a correlation between the presence of cytolytic E. faecalis and clinical severity of ALD. Then, a therapeutic effect of bacteriophages specifically targeting this bacterium was found in humanized mice colonized with bacteria from ALD patients [84]. However, the involvement and mechanism of gut virome in conjunction with bacteriome in liver disease warrant further investigation.
In addition to live microorganisms, fractions and extracts from bacteria, which are recently recognized as ‘postbiotics’, have been reported to profoundly affect host health [85]. The newest member of the biotics family, postbiotics, refers to bioactive compounds produced by food-grade microorganisms. Although the mechanisms involved in postbiotics bioactivity have not been fully understood, studies have reported various functions of postbiotics, including anti-inflammatory, antihypertensive and antioxidant activity [86][87][88]. For example, Benjamin at al found that whole-cell lysates of the non-commensal bacterium Methylococcus capsulatus Bath could improve glucose regulation, diminish hepatic immune infiltration, and change the gut microbiome composition in diet-induced obese mice [89]. Several properties of postbiotics, including their clear chemical structures, stability, and safety dose parameters grant them the potential to become the new generation of health products [90][91]. Although postbiotics is an attractive strategy in the therapeutics of liver diseases, further studies into its mechanism and efficacy are still needed.
4. Mechanism of Action of Microbiome in Liver Disease Pathogenesis and Development
The relationship between dysbiosis of gut microbiome and various liver diseases has been demonstrated in both clinical and preclinical studies. Understanding the mechanism underlying how gut-liver axis functions in relation to liver disease has also been the focus of numerous research studies [1]. Such studies highlight therapeutic approaches of modulating microbiota to treat liver diseases. Although different microbes may contribute to certain types or stages of liver diseases, several common pathways had been discovered. Nutrients and metabolites derived from commensal gut bacterial reach the liver through the portal vein, which can affect hepatocytes metabolism and cause liver damage [1][2][92]. Bacterial and bacterial-derived products, such as lipopolysaccharides, have been implicated in the development of local and systemic immunity, and contribute to liver diseases by interrupting the gut barrier and stimulating liver inflammation [93][94][95][96]. Here we summarize the current knowledge of how gut microbiome affect liver diseases in two aspects, metabolism and immunity (Figure 2). More importantly, bacterial derived metabolites act as crucial modulators of both innate and adaptive immunity [11][97][98]. The microbial metabolites or components implicated in liver diseases are summarized in Table 2.
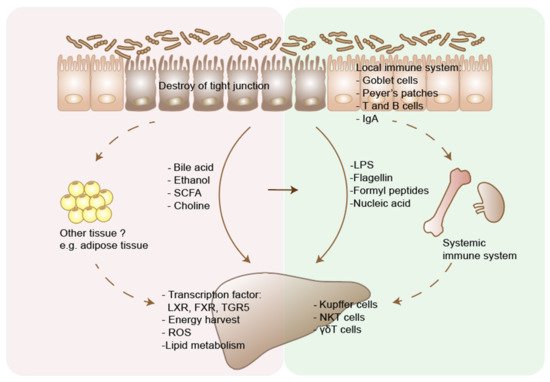
Figure 2. Two main pathways through which gut microbiome affects liver disease. Liver disease results in dysbiosis and intestinal bacterial overgrowth. Disturbed bile acid metabolism affects liver metabolism through regulating the transcription factors, including FXR, LXR, TGR5. Microbiota promote host energy harvest through increasing short chain fatty acids (SCFAs) production. Dietary and microbiota produced ethanol and its metabolites cause hepatocytes damage through generating reactive oxygen species (ROS). Conversion of choline by microbiota causes choline deficiency in host body, which then disturbs lipid metabolism in liver. Gut microbiota could also affect the metabolism of other tissues, e.g., the adipose tissue, and indirectly affect liver diseases, through the chemokines or cytokines interaction. On the other hand, increased intestinal permeability in liver disease leads to translocation of bacteria and microbial products, including LPS, flagellin, formyl peptides and nuclear acids. These pathogens associated molecular patterns (PAMPs) are recognized by pattern recognition receptors (PRRs), such as Toll-like receptors and formyl-peptide receptors, and cause immune cell response in the liver. Microbiota could also modulate host immunity through affecting both local and systemic immune system, which may indirectly affect the progression of liver disease. In addition, microbial metabolites can interact with immune system in liver disease. Primary bile acid could induce NKT cell accumulation in the liver and decrease tumor growth. LXR, liver X receptors; FXR, farnesoid X receptor; G-protein-coupled bile acid receptor 5 (TGR5).
Table 2. Microbial metabolites or components implicated in liver diseases.
| Microbial Metabolites or Components | Function of Microbiota | Effects | Mechanisms | References |
|---|---|---|---|---|
| Bile acids | Microbiota deconjugates primary bile acids and changes the primary/secondary ratio of bile acids. | Effects in the gut | Tauro-conjugated beta-muricholic acids (TβMCA) inhibit FXR-dependent Fgf15 expression in the ileum, which then increase bile acid synthesis in the liver. | [99] |
| Effects In the liver | Bile acids activate FXR, vitamin D receptor, and TGR5, and regulate the metabolism of glucose, fatty acid, triglyceride and VLDL. | [100] | ||
| Effects on immune system | Primary bile acids regulate CXCL16 level on liver sinusoidal endothelial cells, which controls the NKT cells accumulation and inhibit liver tumor growth. | [11] | ||
| Short chain fatty acids | Microbiota produce short chain fatty acids through fermentation of polysaccharides | Effects in the gut | Butyrates are energy sources for enterocytes and help maintain the integrity of the intestinal barrier. | [101] |
| SCFAs act on G-protein receptors (GPCRs) GPR41 and GPR43 on gut enteroendocrine L cells and enhance nutrient absorption. | [102] | |||
| Effects In the liver | SCFAs could increase hepatic lipid oxidation and lower hepatic lipid synthesis. | [103] | ||
| Effects on immune system | Butyrates suppress inflammation through inducing the differentiation of colonic Treg cells. | [104] | ||
| Ethanol | Microbiota contribute to the metabolism of ethanol into acetaldehyde and acetate. Microbiota also produce ethanol through fermentation of carbohydrates. |
Effects in the gut | Acetaldehyde could significantly increase intestine permeability. | [55] |
| Effects In the liver | Alcohol and its metabolites cause damage to hepatocytes through generation of free radicals, which cause oxidative stress. | [105] | ||
| Effects on immune system | Ethanol-induced gut barrier dysfunction and translocation leads to activation of Kupffer cells, infiltrating neutrophils and macrophages, which release proinflammatory cytokines and cause parenchymal cell death. | [106] | ||
| Choline | Choline can be processed to trimethylamine (TMA) by intestinal bacteria, which can lead to reduced availability of dietary choline. | Effects In the liver | Choline deficiency inhibits VLDL excretion from the liver and leads to the hepatic accumulation of triglycerides. | [107] |
| Trimethylamine N-oxide (TMAO), derived from TMA, is associated with liver damage. However, a causal relationship between the two still needs to be clarified. | [108] | |||
| Pathogen-associated molecular patterns, including cell wall components and DNA | LPS is the cell component of gram-negative bacteria. Flagellin, the primary structural component of flagella. |
Effects in the gut immune system | Activation of TLRs on intestinal epithelial cells promotes epithelial cell proliferation, secretion of IgA and antimicrobial peptides. | [109] |
| Effects In the liver immune system | Activation of TLRs on hepatic Kupffer cells and HSCs leads to inflammation and fibrosis, through inducing a range of cytokines, including IL-1, IL-6 and TNF. | [110] |
4.1. Metabolism
4.1.1. Bile Acids
Gut microbiota are closely associated with bile acids synthesis and metabolism in the host, contributing to liver diseases by modulating the pool and functionality of bile acids (BAs), while the microbial composition could also be modulated by bile acids.
Bile acids are synthesized from cholesterol by a series of oxidative transformations in the liver and secreted in the form of primary BAs and metabolized in the intestine into secondary BAs by the microbiome. Firstly, microbiota has been found to play a role in the regulation of bile acid synthesis, which is a complex process, catalyzed by a series of enzymes. Study discovered that several of these enzymes, including enzymes involved in bile acid synthesis, CYP7A1, CYP7B1, and CYP27A1, were under microbial regulation [99]. Secondly, microbiota metabolize the BAs in the gut. In humans, the primary bile acids are chenodeoxycholic acid (CDCA) and cholic acid (CA), while in rodents are CA and muricholic acids (MCAs), mainly beta-MCA. Approximately 95% of primary conjugated bile acids are reabsorbed from the intestine by the apical sodium dependent bile acid transporter (ASBT, or known as IBAT), and recirculated back to liver via the portal vein. Through this process, the bile acids form a circle between the liver and the gut, which is called enterohepatic circulation. The microbiota plays an important role in this circulation, affecting BA metabolism. BAs are deconjugated by the bacteria expressing bile salt hydrolase (BSH). Metagenomic analyses reveals a high enrichment of BSH in gut microbiota comparing with other environmental metagenomes, akin to the ones from sea and soil [111]. Deconjugated primary bile acids were metabolized into secondary bile acids deoxycholic acid (DCA) and lithocholic acid (LCA) through 7-dehydroxylation. This process relies on the bacteria with bile acid-inducible (bai) genes, which have been identified in the genera Clostridium and Eubacterium, both belonging to the Firmicutes phylum [92][112]. In the absence of these bacteria, primary conjugated bile acids dominate the bile acid pool [113][114][115]. GF mice have higher proportion of MCAs in the liver, which is a primary bile acid in mice. Apart from DCA and LCA, two major secondary bile acids, ~50 other secondary bile acids exist in human feces, among which isoDCA and isoLCA are the second most abundant secondary bile acids. isoDCA has less detergent capacity and toxicity than DCA and is produced by E. lentum, C. perfringens, and R. gnavus. [116][117][118] The conversion of DCA to isoDCA favors the growth of the keystone genus Bacteroides [116].
Bile acids can affect host metabolism through acting as ligands for a variety of transcription factors, the alteration of which involves in metabolic and hepatic disease, including the FXR, LXR, the TGR5, and the vitamin D receptor [92]. Also, these functions of bile acids are closely related with gut microbiota functionality [92]. The gut microbiota promotes FXR signaling in mice through deconjugating TβMCA [99], a natural antagonist of FXR. The gut microbiota is also necessary for producing secondary bile acids which are ligands for TGR5 [119]. Both FXR and TGR5 act as modulators of various components in glucose, lipid and energy metabolisms. Intrahepatic retention of hydrophobic BAs was increased in a NASH-HCC mouse model, which were closely associated with gut microbiota alterations [120]. Obesity related alterations of gut microbiota increased the levels of DCA in mice. The enterohepatic circulation of DCA was implicated in the development of obesity-associated hepatocellular carcinoma, through provoking senescence-associated secretory phenotype in hepatic stellate cells in mice [70]. DCA and LCA have also been demonstrated as a carcinogen in colon cancer both in mouse models and cell lines [121][122][123]. In contrast, ursodeoxycholic acid (UDCA) protects against colon cancer [124]. Moreover, bile acid could largely affect the composition of gut microbiota. Biliary obstruction leads to bacterial overgrowth and translocation of Gram-negative aerobic population, which may perpetuate systemic sepsis [125]. Data also suggest that dysbiosis in cirrhotic patients might be partially due to low bile acid input [126]. In summary, change in microbiota can affect liver disease through the regulation of bile acid metabolism.
4.1.2. Short Chain Fatty Acids
Short Chain Fatty Acids (SCFAs), including acetic, propionic, and butyric acid, are produced by intestinal microbiota through fermentation of undigested carbohydrates. These SCFAs affect host immunity and metabolism in various ways [127]. Gut microbiome, which consume dietary carbohydrate, affect synthesis of SCFAs and contribute to liver homeostasis [128].
Butyrate is an energy source for enterocytes and plays central metabolic roles in maintaining the function of the intestinal barrier [129][130]. A reduction in butyrate is associated with decrease of intestinal tight junction and increased permeability [131]. Supplementation of tributyrin, a glycerol ester of butyrate, reduced intestinal permeability and ameliorated liver injury in an alcohol-induced liver disease mouse model [131]. Besides, studies found that SCFAs can regulate energy harvest and expenditure in the liver, peripheral adipose tissue and muscle [103][132][133]. Previous data found an association between obesity and enrichment of genes involved in carbohydrate fermentation [134]. In contrast, several animal studies found that oral administration of SCFAs reduced body weight in obese mice, mainly through increasing energy expenditure [103][135]. In humans, an increased content of butyrate-producing bacteria was found in recipients with metabolic syndrome receiving intestinal microbiota transplantation from lean donors [136]. And this increase of butyrate-producing bacteria is associated with an improve in insulin sensitivity. The mechanism of how SCFAs regulate energy harvest could be related to the G-protein coupled receptors (GPCRs) of gut enteroendocrine L cells. Activation of these receptors could contribute to the secretion of satiety hormones PYY and GLP-1 [137]. However, the delay in gut transit might also enable increased nutrient absorption. Thus, further human studies on the metabolic function of SCFA producing bacteria should be performed.
4.1.3. Ethanol
Alcohol is absorbed by stomach and small intestine and reaches the liver via the portal vein. Alcohol and its metabolites cause damage to hepatocytes through generation of free radicals, especially reactive oxygen species (ROS). Besides the direct effect on the liver, ethanol and its metabolites also destroy tight junction of intestine cell wall [138]. Intestinal microbiome has been discovered to mediate the increase in intestinal permeability and liver damage induced by ethanol.
Gut microbiota and enterocytes metabolize the ethanol into acetaldehyde and acetate [139][140]. Plasma concentrations of ethanol were increased, while luminal concentrations of acetaldehyde were decreased in antibiotic-treated rats than in controls, after acute alcohol intake. In vitro analysis discovered that acetaldehyde but not ethanol could significantly increase intestine permeability measured by dextran flux, suggesting the importance of the microbiome relied on the oxidation of ethanol into acetaldehyde in gut barrier alteration [55]. Acetaldehyde has also been demonstrated to disrupt intestinal tight junction and increase permeability in epithelial cell line Caco-2 cells [141]. With disturbed gut barrier, pathogen associated molecular pattern (PAMPs) reached the liver and contributed to liver damage and inflammation [142]. Sterilization with antibiotics prevented liver injury in rats following long-term exposure to ethanol [143]. In contrast, greater liver injury and inflammation were observed in germ-free mice after oral consumption of ethanol compared to conventional mice [139]. The discrepancy might be caused by different duration of ethanol exposure. While reducing the intestinal bacterial load with antibiotics attenuated chronic liver inflammation, the complete absence of the microbiota also causes problems in face of acute injury.
In addition to oral consumption, ethanol can also be derived from carbohydrates fermentation by gut microbiota [144]. Similarities of histological features exists between alcoholic and non-alcoholic fatty liver diseases, which suggest that certain common pathogenic processes underpin these two liver disease types. Patients with nonalcoholic fatty liver disease are discovered to have increased luminal and circulation levels of ethanol, with an increase in proteobacteria, which produce alcohol [29]. This provides evidence for non-dietary ethanol produced by intestinal bacteria in the pathogenesis of NASH. Similarly, modest increase in breath ethanol were detected in obese patients compared with lean controls with biopsy proven NASH, who have not drunk alcohol recently [145]. Ob/ob mice that have developed NASH was discovered to have higher early-morning breath alcohol content compared with lean mice [146].
4.1.4. Choline
Choline is an essential nutrient, which is important for cell structure and neurotransmitter synthesis [147]. Mice feed a choline-deficient diet for 4 weeks can induce a NASH-like syndrome and thus be used to model human NASH [148][149]. Choline deficiency has also been correlated with fatty liver in human studies [33][108]. In the liver, choline is metabolized into phosphatidylcholine. Phosphatidylcholine assist the synthesis and excretion of VLDL, and its deficiency leads to hepatic accumulation of triglycerides [107]. In addition, choline can also be processed to trimethylamine (TMA) by intestinal bacteria, which are absorbed through the microvilli and circulated via the portal vein to the liver [150]. In the absence of microbiota, as in germ-free mice and antibiotic-pretreated animals, the amount of urinary TMA was greatly reduced, suggesting an important role for the gut microbiota in choline metabolism [151]. TMA is then converted to trimethylamine N-oxide (TMAO) [152]. An adverse association was found between the plasma TMAO level and the severity of NAFLD in hospital and community-based adults [108]. Besides, increased TMAO was correlated with cardiovascular events both in human and mice [24][153]. Altered intestinal microbiota could stimulate hepatic fat deposition and contribute to liver injury through modulation of dietary choline metabolism [154].
In 129S6 mice, which is susceptible to metabolic diseases, choline metabolism was found as a cause for its disease susceptibility [155]. The bioavailability of choline was reduced in this murine strain as a result of the conversion of choline into methylamines by microbiota. The decrease of circulating plasma choline levels created a phenotype similar to that caused by choline-deficient diets, suggesting an active role of choline metabolism by microbiota in the development of NAFLD [155]. On the other hand, gut microbiota also changed in response to choline deficiency. In individuals fed a choline depletion diet, higher levels of fat accumulation in the liver were associated with the increased levels of Gammaproteobacteria and Erysipelotrichi in the gut, underscoring the role of microbiota in metabolic disorders [33].
4.2. Immunity
4.2.1. The Impact of Commensal Bacterial on Local and Systemic Immunity
Commensal microorganism intestinal colonization is influenced by maternal transfer, mode of delivery, contact and diet [96]. An adult-like intestinal microbiota is established by three years of age and maintained a relatively stable state [156]. However, microbiome can be disturbed in disease states or with administration of antibiotics and medications [157][158][159]. The gut microbiota is involved in the development of local mucosal immunity and regulation of systemic immunity [95][160][161].
The importance of gut microbiota on host mucosal immunity is well corroborated through various studies comparing germ-free mice with conventionally raised mice [162][163]. The intestinal immune system is underdeveloped in germ-free animals. Firstly, germ-free mice have fewer and smaller goblet cells, and thinner mucus layer, which is the first anatomical site of defense against intestinal pathogens [161][163]. Then, germ-free mice have smaller mesenteric lymph nodes and smaller Peyer’s patches [164][165]. Bacteria also helped the genesis of isolated lymphoid follicles (ILFs) in mice and the maturation of ILFs into large B-cell clusters [166]. Finally, T and B cells in the Peyer’s patches, CD4+ lymphocytes in the lamina propra, and IgA-secreting plasma cells are numerically affected by germ-free conditions and some of which can be restored by conventionalization [167][168]. These features render the germ-free animals impaired local immune response to pathogens [169][170][171], and more prone to infections [172].
In addition to mucosal immune response, gut microbiota also plays a vital role in shaping systemic immunity. Early studies demonstrated the role of commensal microbes in the regulation of bone marrow myelopoiesis [95][173][174]. In line with previous studies, recent study reports a global decrease of the proportions and differentiation potential of specific myeloid cell progenitors, granulocyte and/or monocyte progenitors (GMPs) in germ-free mice [175]. Thus, the immune defense against the infection with Listeria monocytogenes were decreased in germ-free and oral-antibiotic-treated mice [175].
Lack of commensal bacteria also increase host susceptibility to allergic disease through increasing serum IgE and circulating basophil populations [160]. In addition, a single commensal bacterial species, segmented filamentous bacteria, can drive autoimmune arthritis in germ free animals partially through inducing autoantibodies production [176]. These evidence show the ability of microbiota to shape the global immune system and modulate the host susceptibility to inflammation and infection.
4.2.2. The Impact of Gut Microbiome and Its Derivatives on Innate and Adaptive Immunity
Innate Immunity
Microorganisms and their metabolic products are implicated in the homeostasis of the innate immune system and can also be regulated by the host responses [177].
Innate immune system locates at the host-microbiome interface to protect the integrity of gut barrier [177]. The intestinal epithelial cells are equipped with pattern recognition receptors (PRRs), such as Toll like receptors (TLRs) and NOD-like receptors (NLRs), which can sense conserved components of the microbiome. The lack of these innate immune receptors leads to dysbiosis and disrupted gut epithelial barrier, predisposing tissue to inflammation [178][179]. The inflammasome signaling in epithelial cells, which could induce IL-18 and downstream antimicrobial peptides production, is the best-characterized molecular mechanisms for maintaining epithelial integrity [180]. The microbiota-associated metabolite taurine has been found to positively regulate the NLRP6 inflammasome pathway, while histamine and spermine play a negative role in this situation [181]. What is more, SCFAs can activate the NLRP3 inflammasome pathway through binding to GPR43 and GPR109A on epithelial cells and protect against colitis [180]. Besides metabolites, the protozoan Tritrichomonas musculis has been found to activate the inflammasome to protect against bacterial infections, at the same time increasing the risk of inflammatory disease [182].
In addition to epithelial cells, microbiota and their metabolites also interact with innate lymphoid cells and other non-classical lymphocytes. First, innate lymphoid cells are a newly discovered arm of the innate immune system, which play critical roles in mucosal immunity [183]. Genome-wide chromatin and transcriptional profiling combined with single-cell transcriptomic analysis described a comprehensive map of ILCs subsets in the intestine and found a critical impact of microbiota on the gene expression of ILCs, which is involved in the cell fate determination [183]. Again, metabolites from the microbiota have been found to regulate ILCs function. Zelante et al. discovered that tryptophan metabolites can induce IL-22 production of ILCs, which in turn regulates the survival of microbial communities, providing resistance to fungus infection [98]. Next, liver-resident γδT cells were identified and demonstrated to be a connection between microbiota and hepatic immune response. Hepatic γδT cells can be activated by microbiota lipid antigens and produce proinflammatory cytokine IL-17A [184]. Finally, NKT cells are enriched in the liver and lies in the middle of the interactions between microbiota and hepatic immunosurveillance. Primary bile acid can increase CXCL16 expression in liver sinusoidal endothelial cells, which induce NKT cell accumulation and decrease liver tumor growth. Colonization of bile acid-metabolizing bacteria reversed both the NKT cell accumulation and its effect on tumor inhibition [11].
Adaptive Immunity
Microbiota can influence the adaptive immune system in the gut mucosa, such as differentiation of CD4+ T cells and IgA-producing B cells in Peyer’s patches and lamina propria. Imbalances in the gut microbiota triggers immune disorders, which can result in systemic outcomes distant from the site of colonization [185].
IgA is secreted in the gut and serves as an important first-line barrier restricting the access of intestinal antigens to the host. Lack of IgA changes the function of microbes in intestine [186]. On the contrary, microbial stimulation is required for the production of IgA, as demonstrated in the example of segmented filamentous bacterium (SFB) and Alcaligenes [164][187]. The production of IgA can be regulated by gut microbiota in both T-cell-dependent and T-cell-independent pathways. T-cell-dependent responses are usually directed against protein antigens [188]. T-cell-independent response is elicited by commensal bacteria and produce polyreactive specificities with low affinity for commensals [189][190], while limited exceptions exist [191]. On the other hand, some bacteria have been found to degrade the secretory component of IgA and IgA itself [192]. IgA coating allows bacteria translocation [193]. An enrichment of IgA-coated Escherichia coli was found in Crohn’s disease-associated peripheral spondyloarthritis compared to Crohn’s disease alone. Colonization of this Escherichia coli induced T helper 17 cell immunity and lead to more severe colitis and arthritis in mouse models [194].
CD4+T cells mainly locate in the lamina propria, which differentiate into Tregs and various T helper cells upon activation by microbiota. Th17 cells and Treg cells are the most extensively studied subsets of T cells in the interaction of microbiota and host diseases [96]. Intestinal microbes, especially SFB, can induce intestinal Th17 cells response [195]. Th17 cells have been found enriched in the intestine of human IBD patients [194]. The transplantation of IBD microbiota into germ-free mice increased the amount of intestinal Th17 cells and Th2 cells, while decreased the number of a specific Treg cells, the ROR γt+ Treg cells [196]. The imbalance between Th17 cells and RORγt+ Treg cells also accounted for disease severity in the colitis mouse model [196]. Next, the number and function of Treg cells can be affected by gut microbiota [197]. For instance, species of 73 genus [198], Bacteroides fragilis and its polysaccharide A (PSA) [199], promoted Treg cell accumulation and differentiation. What is more, inoculation of Clostridium and oral treatment of PSA can lead to resistance or even reversing of experimental colitis in mice [198][199]. Finally, microbiota metabolites, SCFA, have been discovered to regulate the size and function of Treg cell pool and protect against colitis in mouse models [200]. Later, SCFAs was also found to directly promote T cell differentiation into both effector and regulatory T cells [201]. These evidence reveal that microbiota and its metabolites underlie adaptive immune regulation and promote colonic homeostasis and health.
5. Gut Microbial Molecules in Liver Immune Modulation
Microbial components translocate through the portal vein into the liver and directly modulate the immune response in liver, thus affecting its pathogenesis, progression, and development. The immune system recognizes pathogen-associated molecular patterns (PAMPs) through pattern recognition receptors (PRRs). LPS is the cell component of gram-negative bacteria, which can increase inflammation, metabolic syndrome, and fibrosis in the liver [202][203]. NASH patients had higher levels of LPS in peripheral circulation and in the liver compared with controls [204]. In high fat diet fed mice, the level of LPS increases by 2–3 folds [202], with an increase of LPS-containing microbiota. On the other hand, LPS infusion alone can increase visceral and subcutaneous fat deposition in mice which is similar to the effect induced by high-fat diet [202]. The number of macrophages in adipose tissue and levels of inflammatory markers were also increased due to LPS infusion [202]. Most of these features, both in LPS and high-fat diet-induced metabolic diseases, were abolished or attenuated in the CD14 mutant mice, which is a main LPS receptor [202].
LPS stimulates Kupffer cells through an essential signaling cascade and thus induces the production of immune regulating cytokines [93]. After integrating with LBP, the LPS-LBP complex binds to CD14 which associates with TLR on the cell surface and stimulates the downstream mitogen-activated protein kinases (MAPK), JNK, p38, and NF-κB pathway. This leads to the transcription of proinflammatory cytokines, including TNF, IL-1, and IL-6. Evidence from animal models and patients support that this signaling pathway is activated in NASH. As mentioned above, mice without CD14 were protected from the development of steatosis after LPS treatment [202]. In MCD-diet fed mice, TLR4 ligand challenge aggravated liver injury and increases proinflammatory cytokine secretion [205].
Other microbial components, flagellin, formyl peptides, and nucleic acid can also regulate immune response and impact liver pathophysiology. Flagellin, the primary structural component of flagella, is a typical pathogen-associated molecular pattern. It can be sensed by TLR5 on the cell surface and signals through MyD88, resulting in the production of inflammatory cytokines and chemokines [206]. Flagellin administration can induce liver injury, which is associated with neutrophils and macrophages accumulation in the liver [207]. Next, human Formyl-peptide receptors FPRs is considered as an PRR, which recognize the peptides cleaved of bacterial [208]. Fprs deficiency exacerbated the severity of infection in mice, which is related with impaired neutrophil recruitment to the liver [209]. This suggests a role of formyl-peptide in the induction of immune response in liver in the early stage of infection. Finally, nucleic acids from bacteria also play a major role in intestinal immune homeostasis. Unmethylated cytosine phosphate guanosine (CpG) dinucleotides limit Treg cell’s suppressive function [210], and DNA derived from conventional gut flora DNA (gfNDA) could also inhibit Treg cell conversion and modulate the Treg/Teff equilibrium [169]. TLR-9 recognizes the CpG containing DNA from bacteria and virus. TLR-9 deficient mice showed reduced liver injury, with dampened hepatic neutrophil infiltration [211]. Through these different mechanisms, bacteria and its components modulate the immune balance of the liver against pathogens and antigens.
6. Microbiota in Therapeutics of Liver Diseases
Based on the important roles of dysbiosis in the development of liver diseases, various approaches to restructuring the gut microbiota have been tried in the treatment and prevention of liver diseases. (Figure 3).
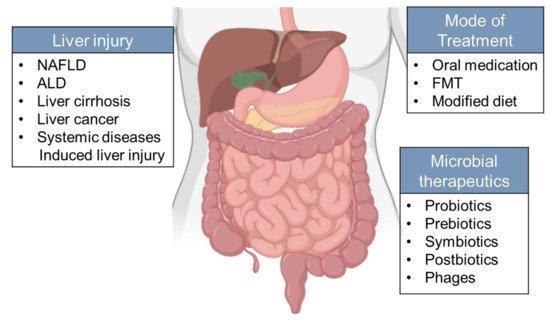
Figure 3. Microbiota-oriented interventions can be applied to different types of chronic liver disease. Strategies aimed at changing gut microbiota include supplementation of prebiotics, probiotics, symbiotics, and postbiotics on the host. However, the therapeutic potential of microorganisms from other kingdoms (phages) remains to be explored. Mode of treatment include oral administration, FMT and modified diet.
Treatments of antibiotics [212], prebiotics [213], and probiotics [214][215] have been demonstrated to hinder the development of NAFLD in mouse models. The underlying mechanisms of these approaches cater for protecting against the increased translocation of bacterial endotoxin and the subsequent activation of Kupffer cells and induction of TNF-α. For example, administration of VSL#3, a multistrain cocktail composed of several species of Lactobacillus and Bifidobacteria, and Streptococcus thermophilus, could decrease liver TNF-α levels and limit inflammatory liver damage in rats receiving HFD [216]. Lactobacillus casei Shirota treatment attenuated the activation of TLR4 and protected against NAFLD in mouse model of fructose-induced steatosis [215]. Then, oligofructose (OFS) supplementation in NASH patients significantly decreased serum ALT and AST levels compared to placebo in a small-scale pilot study [217], suggesting the potential of prebiotics in the treatment of NASH. Finally, the combination of probiotics and prebiotics also showed positive results in the treatment of NASH [218].
In cirrhosis, the modulation of gut microbiota has also been demonstrated to be a potential therapy in both preclinical experiments and clinical trials. VSL#3 treatment significantly improved the Child–Turcotte–Pugh score and reduced the need for hospitalization in cirrhotic patients compared with placebo-treated controls [219]. In addition, FMT increased gut microbiota diversity and beneficial taxa, and improved the cognition levels in patients with recurrent HE, compared with patients receiving standard of care alone [220]. FMT showed similar protective effects in carbon tetrachloride (CCl4)-induced HE in rats [221].
Recently, the potential role of gut microbiota in the chemotherapy and immunotherapy has been highlighted in several studies. The loss of certain commensal bacterial by antibiotics treatment inhibited anti-tumor Th17 cell induction, and thus compromised the efficacy of cyclophosphamide in the treatment of sarcomas in mice [72]. Antibiotics also inhibited the efficacy of anti-CTLA4 immunotherapy in mouse sarcomas, possibly through to the decrease of B. fragilis and B.thetaiotaomicron which could induce the immune response mediated by Th1 cells [222]. In another mouse model of melanoma, commensal bacterial enhanced the efficiency of programmed cell death protein 1 ligand (PD-L1) therapy, which has been identified to be the effect of Bifidobacterium [223]. However, it remains to be studied whether the beneficial anti-tumor properties of gut microbiota can be applied to liver cancer treatment.
In addition to bacteria, dysbiosis of microorganisms from other kingdoms, including fungi, archaea and viruses, and postbiotics were found to be associated with liver diseases [78][79][80]. An antifungal drug has been found to prevent ethanol-induced steatohepatitis in mice [79]. However, whether manipulating the intestinal mycobiome can be an effective strategy for alcohol-related liver disease still need to be tested in patients. Bacteriophages are viruses which can infect and kill bacteria and play a role in preserving the microbiome equilibrium. A recent study found that bacteriophages specifically targeting cytolytic E. faecalis can abolish ethanol-induced liver disease in humanized mice [84]. Still, clinical trials are warranted to validate the therapeutic potential of such phage-oriented strategy.
This entry is adapted from the peer-reviewed paper 10.3390/microorganisms9051017
References
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411.
- Schnabl, B.; Brenner, D.A. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014, 146, 1513–1524.
- Tilg, H.; Cani, P.D.; Mayer, E.A. Gut microbiome and liver diseases. Gut 2016, 65, 2035–2044.
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M.; et al. The gut microbiota and host health: A new clinical frontier. Gut 2016, 65, 330–339.
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171.
- Dapito, D.H.; Mencin, A.; Gwak, G.Y.; Pradere, J.P.; Jang, M.K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516.
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775.
- Ponziani, F.R.; Bhoori, S.; Castelli, C.; Putignani, L.; Rivoltini, L.; Del Chierico, F.; Sanguinetti, M.; Morelli, D.; Paroni Sterbini, F.; Petito, V.; et al. Hepatocellular Carcinoma Is Associated With Gut Microbiota Profile and Inflammation in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 107–120.
- Dubinkina, V.B.; Tyakht, A.V.; Odintsova, V.Y.; Yarygin, K.S.; Kovarsky, B.A.; Pavlenko, A.V.; Ischenko, D.S.; Popenko, A.S.; Alexeev, D.G.; Taraskina, A.Y.; et al. Links of gut microbiota composition with alcohol dependence syndrome and alcoholic liver disease. Microbiome 2017, 5, 141.
- Hoyles, L.; Fernandez-Real, J.M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H.; et al. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1070–1080.
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360.
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011, 54, 133–144.
- Zuo, T.; Zhang, F.; Lui, G.C.Y.; Yeoh, Y.K.; Li, A.Y.L.; Zhan, H.; Wan, Y.; Chung, A.; Cheung, C.P.; Chen, N.; et al. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology 2020.
- Phipps, M.M.; Barraza, L.H.; LaSota, E.D.; Sobieszczyk, M.E.; Pereira, M.R.; Zheng, E.X.; Fox, A.N.; Zucker, J.; Verna, E.C. Acute Liver Injury in COVID-19: Prevalence and Association with Clinical Outcomes in a Large US Cohort. Hepatology 2020.
- Ferrere, G.; Wrzosek, L.; Cailleux, F.; Turpin, W.; Puchois, V.; Spatz, M.; Ciocan, D.; Rainteau, D.; Humbert, L.; Hugot, C.; et al. Fecal microbiota manipulation prevents dysbiosis and alcohol-induced liver injury in mice. J. Hepatol. 2017, 66, 806–815.
- Liu, Q.; Duan, Z.P.; Ha, D.K.; Bengmark, S.; Kurtovic, J.; Riordan, S.M. Synbiotic modulation of gut flora: Effect on minimal hepatic encephalopathy in patients with cirrhosis. Hepatology 2004, 39, 1441–1449.
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e1055.
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845.
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846.
- Wieland, A.; Frank, D.N.; Harnke, B.; Bambha, K. Systematic review: Microbial dysbiosis and nonalcoholic fatty liver disease. Aliment Pharmacol. Ther. 2015, 42, 1051–1063.
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546.
- Perry, R.J.; Peng, L.; Barry, N.A.; Cline, G.W.; Zhang, D.; Cardone, R.L.; Petersen, K.F.; Kibbey, R.G.; Goodman, A.L.; Shulman, G.I. Acetate mediates a microbiome-brain-beta-cell axis to promote metabolic syndrome. Nature 2016, 534, 213–217.
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60.
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63.
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266.
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023.
- Schwiertz, A.; Taras, D.; Schafer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195.
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G.; et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 2016, 535, 376–381.
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609.
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127.
- Wong, V.W.; Tse, C.H.; Lam, T.T.; Wong, G.L.; Chim, A.M.; Chu, W.C.; Yeung, D.K.; Law, P.T.; Kwan, H.S.; Yu, J.; et al. Molecular characterization of the fecal microbiota in patients with nonalcoholic steatohepatitis—A longitudinal study. PLoS ONE 2013, 8, e62885.
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G.; et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 1172.
- Spencer, M.D.; Hamp, T.J.; Reid, R.W.; Fischer, L.M.; Zeisel, S.H.; Fodor, A.A. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 2011, 140, 976–986.
- Raman, M.; Ahmed, I.; Gillevet, P.M.; Probert, C.S.; Ratcliffe, N.M.; Smith, S.; Greenwood, R.; Sikaroodi, M.; Lam, V.; Crotty, P.; et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2013, 11, 868.e861–875.e863.
- Backhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984.
- Rabot, S.; Membrez, M.; Bruneau, A.; Gerard, P.; Harach, T.; Moser, M.; Raymond, F.; Mansourian, R.; Chou, C.J. Germ-free C57BL/6J. mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J. 2010, 24, 4948–4959.
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723.
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031.
- Le Roy, T.; Llopis, M.; Lepage, P.; Bruneau, A.; Rabot, S.; Bevilacqua, C.; Martin, P.; Philippe, C.; Walker, F.; Bado, A.; et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2013, 62, 1787–1794.
- Mutlu, E.A.; Gillevet, P.M.; Rangwala, H.; Sikaroodi, M.; Naqvi, A.; Engen, P.A.; Kwasny, M.; Lau, C.K.; Keshavarzian, A. Colonic microbiome is altered in alcoholism. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G966–G978.
- Chen, Y.; Yang, F.; Lu, H.; Wang, B.; Chen, Y.; Lei, D.; Wang, Y.; Zhu, B.; Li, L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 2011, 54, 562–572.
- Bajaj, J.S.; Heuman, D.M.; Hylemon, P.B.; Sanyal, A.J.; White, M.B.; Monteith, P.; Noble, N.A.; Unser, A.B.; Daita, K.; Fisher, A.R.; et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 2014, 60, 940–947.
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64.
- Caussy, C.; Tripathi, A.; Humphrey, G.; Bassirian, S.; Singh, S.; Faulkner, C.; Bettencourt, R.; Rizo, E.; Richards, L.; Xu, Z.Z.; et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat. Commun. 2019, 10, 1406.
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Primers 2018, 4, 16.
- Vassallo, G.; Mirijello, A.; Ferrulli, A.; Antonelli, M.; Landolfi, R.; Gasbarrini, A.; Addolorato, G. Review article: Alcohol and gut microbiota—The possible role of gut microbiota modulation in the treatment of alcoholic liver disease. Aliment Pharmacol. Ther. 2015, 41, 917–927.
- Bode, J.C.; Bode, C.; Heidelbach, R.; Durr, H.K.; Martini, G.A. Jejunal microflora in patients with chronic alcohol abuse. Hepatogastroenterology 1984, 31, 30–34.
- Tuomisto, S.; Pessi, T.; Collin, P.; Vuento, R.; Aittoniemi, J.; Karhunen, P.J. Changes in gut bacterial populations and their translocation into liver and ascites in alcoholic liver cirrhotics. BMC Gastroenterol. 2014, 14, 40.
- Yan, A.W.; Fouts, D.E.; Brandl, J.; Starkel, P.; Torralba, M.; Schott, E.; Tsukamoto, H.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2011, 53, 96–105.
- Canesso, M.C.; Lacerda, N.L.; Ferreira, C.M.; Gonçalves, J.L.; Almeida, D.; Gamba, C.; Cassali, G.; Pedroso, S.H.; Moreira, C.; Martins, F.S.; et al. Comparing the effects of acute alcohol consumption in germ-free and conventional mice: The role of the gut microbiota. BMC Microbiol. 2014, 14, 240.
- Chen, P.; Schnabl, B. Host-microbiome interactions in alcoholic liver disease. Gut Liver 2014, 8, 237–241.
- Bjarnason, I.; Peters, T.J.; Wise, R.J. The leaky gut of alcoholism: Possible route of entry for toxic compounds. Lancet 1984, 1, 179–182.
- Keshavarzian, A.; Holmes, E.W.; Patel, M.; Iber, F.; Fields, J.Z.; Pethkar, S. Leaky gut in alcoholic cirrhosis: A possible mechanism for alcohol-induced liver damage. Am. J. Gastroenterol. 1999, 94, 200–207.
- Rao, R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 2009, 50, 638–644.
- Ferrier, L.; Berard, F.; Debrauwer, L.; Chabo, C.; Langella, P.; Bueno, L.; Fioramonti, J. Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am. J. Pathol. 2006, 168, 1148–1154.
- Keshavarzian, A.; Choudhary, S.; Holmes, E.W.; Yong, S.; Banan, A.; Jakate, S.; Fields, J.Z. Preventing gut leakiness by oats supplementation ameliorates alcohol-induced liver damage in rats. J. Pharmacol. Exp. Ther. 2001, 299, 442–448.
- Keshavarzian, A.; Farhadi, A.; Forsyth, C.B.; Rangan, J.; Jakate, S.; Shaikh, M.; Banan, A.; Fields, J.Z. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J. Hepatol. 2009, 50, 538–547.
- Kirpich, I.A.; Solovieva, N.V.; Leikhter, S.N.; Shidakova, N.A.; Lebedeva, O.V.; Sidorov, P.I.; Bazhukova, T.A.; Soloviev, A.G.; Barve, S.S.; McClain, C.J.; et al. Probiotics restore bowel flora and improve liver enzymes in human alcohol-induced liver injury: A pilot study. Alcohol 2008, 42, 675–682.
- Philips, C.A.; Pande, A.; Shasthry, S.M.; Jamwal, K.D.; Khillan, V.; Chandel, S.S.; Kumar, G.; Sharma, M.K.; Maiwall, R.; Jindal, A.; et al. Healthy Donor Fecal Microbiota Transplantation in Steroid-Ineligible Severe Alcoholic Hepatitis: A Pilot Study. Clin. Gastroenterol. Hepatol. 2017, 15, 600–602.
- Grander, C.; Adolph, T.E.; Wieser, V.; Lowe, P.; Wrzosek, L.; Gyongyosi, B.; Ward, D.V.; Grabherr, F.; Gerner, R.R.; Pfister, A.; et al. Recovery of ethanol-induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut 2018, 67, 891–901.
- Ahluwalia, V.; Betrapally, N.S.; Hylemon, P.B.; White, M.B.; Gillevet, P.M.; Unser, A.B.; Fagan, A.; Daita, K.; Heuman, D.M.; Zhou, H.; et al. Impaired Gut-Liver-Brain Axis in Patients with Cirrhosis. Sci. Rep. 2016, 6, 26800.
- Yanguas, S.C.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; van den Bossche, B.; de Oliveira, C.; Andraus, W.; Alves, V.A.F.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2016, 90, 1025–1048.
- Fouts, D.E.; Torralba, M.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Bacterial translocation and changes in the intestinal microbiome in mouse models of liver disease. J. Hepatol. 2012, 56, 1283–1292.
- Brenner, D.A.; Paik, Y.H.; Schnabl, B. Role of Gut Microbiota in Liver Disease. J. Clin. Gastroenterol. 2015, 49 (Suppl. 1), S25–S27.
- Hugenholtz, F.; de Vos, W.M. Mouse models for human intestinal microbiota research: A critical evaluation. Cell Mol. Life Sci. 2018, 75, 149–160.
- Walter, J.; Armet, A.M.; Finlay, B.B.; Shanahan, F. Establishing or Exaggerating Causality for the Gut Microbiome: Lessons from Human Microbiota-Associated Rodents. Cell 2020, 180, 221–232.
- Fessler, J.; Matson, V.; Gajewski, T.F. Exploring the emerging role of the microbiome in cancer immunotherapy. J. Immunother. Cancer 2019, 7, 108.
- Grat, M.; Wronka, K.M.; Krasnodebski, M.; Masior, L.; Lewandowski, Z.; Kosinska, I.; Grat, K.; Stypulkowski, J.; Rejowski, S.; Wasilewicz, M.; et al. Profile of Gut Microbiota Associated With the Presence of Hepatocellular Cancer in Patients With Liver Cirrhosis. Transpl. Proc. 2016, 48, 1687–1691.
- Huang, Y.; Fan, X.G.; Wang, Z.M.; Zhou, J.H.; Tian, X.F.; Li, N. Identification of helicobacter species in human liver samples from patients with primary hepatocellular carcinoma. J. Clin. Pathol. 2004, 57, 1273–1277.
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101.
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103.
- Viaud, S.; Saccheri, F.; Mignot, G.; Yamazaki, T.; Daillere, R.; Hannani, D.; Enot, D.P.; Pfirschke, C.; Engblom, C.; Pittet, M.J.; et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013, 342, 971–976.
- Yu, L.X.; Schwabe, R.F. The gut microbiome and liver cancer: Mechanisms and clinical translation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 527–539.
- Bajaj, J.S.; Betrapally, N.S.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; White, M.B.; Unser, A.; Thacker, L.R.; Sanyal, A.J.; Kang, D.J.; et al. Salivary microbiota reflects changes in gut microbiota in cirrhosis with hepatic encephalopathy. Hepatology 2015, 62, 1260–1271.
- Bajaj, J.S.; Hylemon, P.B.; Ridlon, J.M.; Heuman, D.M.; Daita, K.; White, M.B.; Monteith, P.; Noble, N.A.; Sikaroodi, M.; Gillevet, P.M. Colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G675–G685.
- Bajaj, J.S.; Betrapally, N.S.; Hylemon, P.B.; Thacker, L.R.; Daita, K.; Kang, D.J.; White, M.B.; Unser, A.B.; Fagan, A.; Gavis, E.A.; et al. Gut Microbiota Alterations can predict Hospitalizations in Cirrhosis Independent of Diabetes Mellitus. Sci. Rep. 2015, 5, 18559.
- Chen, Y.; Ji, F.; Guo, J.; Shi, D.; Fang, D.; Li, L. Dysbiosis of small intestinal microbiota in liver cirrhosis and its association with etiology. Sci. Rep. 2016, 6, 34055.
- Lloyd-Price, J.; Abu-Ali, G.; Huttenhower, C. The healthy human microbiome. Genome Med. 2016, 8, 51.
- Bajaj, J.S.; Liu, E.J.; Kheradman, R.; Fagan, A.; Heuman, D.M.; White, M.; Gavis, E.A.; Hylemon, P.; Sikaroodi, M.; Gillevet, P.M. Fungal dysbiosis in cirrhosis. Gut 2018, 67, 1146–1154.
- Yang, A.M.; Inamine, T.; Hochrath, K.; Chen, P.; Wang, L.; Llorente, C.; Bluemel, S.; Hartmann, P.; Xu, J.; Koyama, Y.; et al. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Investig. 2017, 127, 2829–2841.
- Lang, S.; Demir, M.; Martin, A.; Jiang, L.; Zhang, X.; Duan, Y.; Gao, B.; Wisplinghoff, H.; Kasper, P.; Roderburg, C.; et al. Intestinal Virome Signature Associated With Severity of Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 159, 1839–1852.
- Jiang, L.; Lang, S.; Duan, Y.; Zhang, X.; Gao, B.; Chopyk, J.; Schwanemann, L.K.; Ventura-Cots, M.; Bataller, R.; Bosques-Padilla, F.; et al. Intestinal Virome in Patients With Alcoholic Hepatitis. Hepatology 2020, 72, 2182–2196.
- Bajaj, J.S.; Sikaroodi, M.; Shamsaddini, A.; Henseler, Z.; Santiago-Rodriguez, T.; Acharya, C.; Fagan, A.; Hylemon, P.B.; Fuchs, M.; Gavis, E.; et al. Interaction of bacterial metagenome and virome in patients with cirrhosis and hepatic encephalopathy. Gut 2020.
- Duan, Y.; Llorente, C.; Lang, S.; Brandl, K.; Chu, H.; Jiang, L.; White, R.C.; Clarke, T.H.; Nguyen, K.; Torralba, M.; et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 2019, 575, 505–511.
- Aguilar-Toalá, J.; Garcia-Varela, R.; Garcia, H.; Mata-Haro, V.; González-Córdova, A.; Vallejo-Cordoba, B.; Hernández-Mendoza, A. Postbiotics: An evolving term within the functional foods field. Trends Food Sci. Technol. 2018, 75, 105–114.
- Haileselassie, Y.; Navis, M.; Vu, N.; Qazi, K.R.; Rethi, B.; Sverremark-Ekström, E. Postbiotic modulation of retinoic acid imprinted mucosal-like dendritic cells by probiotic Lactobacillus reuteri 17938 in vitro. Front. Immunol. 2016, 7, 96.
- Gómez-Guzmán, M.; Toral, M.; Romero, M.; Jiménez, R.; Galindo, P.; Sánchez, M.; Zarzuelo, M.J.; Olivares, M.; Gálvez, J.; Duarte, J. Antihypertensive effects of probiotics Lactobacillus strains in spontaneously hypertensive rats. Mol. Nutr. Food Res. 2015, 59, 2326–2336.
- Xu, R.; Shang, N.; Li, P. In vitro and in vivo antioxidant activity of exopolysaccharide fractions from Bifidobacterium animalis RH. Anaerobe 2011, 17, 226–231.
- Jensen, B.A.H.; Holm, J.B.; Larsen, I.S.; von Burg, N.; Derer, S.; Sonne, S.B.; Paerregaard, S.I.; Damgaard, M.V.; Indrelid, S.A.; Rivollier, A.; et al. Lysates of Methylococcus capsulatus Bath induce a lean-like microbiota, intestinal FoxP3(+)RORgammat(+)IL-17(+) Tregs and improve metabolism. Nat. Commun. 2021, 12, 1093.
- Russo, E.; Giudici, F.; Fiorindi, C.; Ficari, F.; Scaringi, S.; Amedei, A. Immunomodulating Activity and Therapeutic Effects of Short Chain Fatty Acids and Tryptophan Post-biotics in Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 2754.
- Homayouni Rad, A.; Aghebati Maleki, L.; Samadi Kafil, H.; Fathi Zavoshti, H.; Abbasi, A. Postbiotics as Promising Tools for Cancer Adjuvant Therapy. Adv. Pharm. Bull. 2021, 11, 1–5.
- Ridlon, J.M.; Bajaj, J.S. The human gut sterolbiome: Bile acid-microbiome endocrine aspects and therapeutics. Acta Pharm. Sin. B 2015, 5, 99–105.
- Abu-Shanab, A.; Quigley, E.M. The role of the gut microbiota in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 691–701.
- Lambertucci, F.; Arboatti, A.; Sedlmeier, M.G.; Motino, O.; Alvarez, M.L.; Ceballos, M.P.; Villar, S.R.; Roggero, E.; Monti, J.A.; Pisani, G.; et al. Disruption of tumor necrosis factor alpha receptor 1 signaling accelerates NAFLD progression in mice upon a high-fat diet. J. Nutr. Biochem. 2018, 58, 17–27.
- Gorjifard, S.; Goldszmid, R.S. Microbiota-myeloid cell crosstalk beyond the gut. J. Leukoc. Biol. 2016, 100, 865–879.
- Vaikunthanathan, T.; Safinia, N.; Lombardi, G.; Lechler, R.I. Microbiota, immunity and the liver. Immunol. Lett. 2016, 171, 36–49.
- Brestoff, J.R.; Artis, D. Commensal bacteria at the interface of host metabolism and the immune system. Nat. Immunol. 2013, 14, 676–684.
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385.
- Sayin, S.I.; Wahlstrom, A.; Felin, J.; Jantti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyotylainen, T.; Oresic, M.; Backhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235.
- Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Ren, S.; Gil, G.; Dent, P. Bile acids as regulatory molecules. J. Lipid Res. 2009, 50, 1509–1520.
- Clausen, M.R.; Mortensen, P.B. Kinetic studies on colonocyte metabolism of short chain fatty acids and glucose in ulcerative colitis. Gut 1995, 37, 684–689.
- Musso, G.; Gambino, R.; Cassader, M. Obesity, diabetes, and gut microbiota: The hygiene hypothesis expanded? Diabetes Care 2010, 33, 2277–2284.
- Den Besten, G.; Bleeker, A.; Gerding, A.; van Eunen, K.; Havinga, R.; van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngoud, D.J.; et al. Short-Chain Fatty Acids Protect Against High-Fat Diet-Induced Obesity via a PPARgamma-Dependent Switch From Lipogenesis to Fat Oxidation. Diabetes 2015, 64, 2398–2408.
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450.
- Lieber, C.S. Alcoholic fatty liver: Its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 2004, 34, 9–19.
- Hoek, J.B.; Pastorino, J.G. Ethanol, oxidative stress, and cytokine-induced liver cell injury. Alcohol 2002, 27, 63–68.
- Mehedint, M.G.; Zeisel, S.H. Choline’s role in maintaining liver function: New evidence for epigenetic mechanisms. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 339–345.
- Chen, Y.M.; Liu, Y.; Zhou, R.F.; Chen, X.L.; Wang, C.; Tan, X.Y.; Wang, L.J.; Zheng, R.D.; Zhang, H.W.; Ling, W.H.; et al. Associations of gut-flora-dependent metabolite trimethylamine-N-oxide, betaine and choline with non-alcoholic fatty liver disease in adults. Sci. Rep. 2016, 6, 19076.
- Abreu, M.T. Toll-like receptor signalling in the intestinal epithelium: How bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 2010, 10, 131–144.
- Mencin, A.; Kluwe, J.; Schwabe, R.F. Toll-like receptors as targets in chronic liver diseases. Gut 2009, 58, 704–720.
- Jones, B.V.; Begley, M.; Hill, C.; Gahan, C.G.; Marchesi, J.R. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 13580–13585.
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259.
- Kellogg, T.F.; Wostmann, B.S. Fecal neutral steroids and bile acids from germfree rats. J. Lipid Res. 1969, 10, 495–503.
- Selwyn, F.P.; Csanaky, I.L.; Zhang, Y.; Klaassen, C.D. Importance of Large Intestine in Regulating Bile Acids and Glucagon-Like Peptide-1 in Germ-Free Mice. Drug Metab. Dispos. 2015, 43, 1544–1556.
- Wahlstrom, A.; Sayin, S.I.; Marschall, H.U.; Backhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50.
- Devlin, A.S.; Fischbach, M.A. A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat. Chem. Biol. 2015, 11, 685–690.
- Hirano, S.; Masuda, N. Transformation of bile acids by Eubacterium lentum. Appl. Environ. Microbiol. 1981, 42, 912–915.
- Hirano, S.; Masuda, N.; Oda, H.; Mukai, H. Transformation of bile acids by Clostridium perfringens. Appl. Environ. Microbiol. 1981, 42, 394–399.
- Kuipers, F.; Bloks, V.W.; Groen, A.K. Beyond intestinal soap--bile acids in metabolic control. Nat. Rev. Endocrinol. 2014, 10, 488–498.
- Xie, G.; Wang, X.; Huang, F.; Zhao, A.; Chen, W.; Yan, J.; Zhang, Y.; Lei, S.; Ge, K.; Zheng, X.; et al. Dysregulated hepatic bile acids collaboratively promote liver carcinogenesis. Int. J. Cancer 2016, 139, 1764–1775.
- Dermadi, D.; Valo, S.; Ollila, S.; Soliymani, R.; Sipari, N.; Pussila, M.; Sarantaus, L.; Linden, J.; Baumann, M.; Nystrom, M. Western Diet Deregulates Bile Acid Homeostasis, Cell Proliferation, and Tumorigenesis in Colon. Cancer Res. 2017, 77, 3352–3363.
- Pai, R.; Tarnawski, A.S.; Tran, T. Deoxycholic acid activates beta-catenin signaling pathway and increases colon cell cancer growth and invasiveness. Mol. Biol. Cell 2004, 15, 2156–2163.
- Nguyen, T.T.; Ung, T.T.; Kim, N.H.; Jung, Y.D. Role of bile acids in colon carcinogenesis. World J. Clin. Cases 2018, 6, 577–588.
- Jacoby, R.F.; Cole, C.E.; Hawk, E.T.; Lubet, R.A. Ursodeoxycholate/Sulindac combination treatment effectively prevents intestinal adenomas in a mouse model of polyposis. Gastroenterology 2004, 127, 838–844.
- Clements, W.D.; Parks, R.; Erwin, P.; Halliday, M.I.; Barr, J.; Rowlands, B.J. Role of the gut in the pathophysiology of extrahepatic biliary obstruction. Gut 1996, 39, 587–593.
- Kakiyama, G.; Pandak, W.M.; Gillevet, P.M.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; Takei, H.; Muto, A.; Nittono, H.; Ridlon, J.M.; et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J. Hepatol. 2013, 58, 949–955.
- Rodriguez-Carrio, J.; Salazar, N.; Margolles, A.; Gonzalez, S.; Gueimonde, M.; de Los Reyes-Gavilan, C.G.; Suarez, A. Free Fatty Acids Profiles Are Related to Gut Microbiota Signatures and Short-Chain Fatty Acids. Front. Immunol. 2017, 8, 823.
- Sharon, G.; Garg, N.; Debelius, J.; Knight, R.; Dorrestein, P.C.; Mazmanian, S.K. Specialized metabolites from the microbiome in health and disease. Cell Metab. 2014, 20, 719–730.
- Wachtershauser, A.; Stein, J. Rationale for the luminal provision of butyrate in intestinal diseases. Eur. J. Nutr. 2000, 39, 164–171.
- Ziegler, K.; Kerimi, A.; Poquet, L.; Williamson, G. Butyric acid increases transepithelial transport of ferulic acid through upregulation of the monocarboxylate transporters SLC16A1 (MCT1) and SLC16A3 (MCT4). Arch. Biochem. Biophys. 2016, 599, 3–12.
- Cresci, G.A.; Glueck, B.; McMullen, M.R.; Xin, W.; Allende, D.; Nagy, L.E. Prophylactic tributyrin treatment mitigates chronic-binge ethanol-induced intestinal barrier and liver injury. J. Gastroenterol. Hepatol. 2017, 32, 1587–1597.
- Yamashita, H.; Maruta, H.; Jozuka, M.; Kimura, R.; Iwabuchi, H.; Yamato, M.; Saito, T.; Fujisawa, K.; Takahashi, Y.; Kimoto, M.; et al. Effects of acetate on lipid metabolism in muscles and adipose tissues of type 2 diabetic Otsuka Long-Evans Tokushima Fatty (OLETF) rats. Biosci. Biotechnol. Biochem. 2009, 73, 570–576.
- Al-Lahham, S.; Roelofsen, H.; Rezaee, F.; Weening, D.; Hoek, A.; Vonk, R.; Venema, K. Propionic acid affects immune status and metabolism in adipose tissue from overweight subjects. Eur. J. Clin. Investig. 2012, 42, 357–364.
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1241214.
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517.
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojarvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e917.
- Chambers, E.S.; Viardot, A.; Psichas, A.; Morrison, D.J.; Murphy, K.G.; Zac-Varghese, S.E.; MacDougall, K.; Preston, T.; Tedford, C.; Finlayson, G.S.; et al. Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut 2015, 64, 1744–1754.
- Szabo, G.; Mandrekar, P. Focus on: Alcohol and the liver. Alcohol Res. Health 2010, 33, 87–96.
- Chen, P.; Miyamoto, Y.; Mazagova, M.; Lee, K.C.; Eckmann, L.; Schnabl, B. Microbiota Protects Mice Against Acute Alcohol-Induced Liver Injury. Alcohol Clin. Exp. Res. 2015, 39, 2313–2323.
- Hamarneh, S.R.; Kim, B.M.; Kaliannan, K.; Morrison, S.A.; Tantillo, T.J.; Tao, Q.; Mohamed, M.M.R.; Ramirez, J.M.; Karas, A.; Liu, W.; et al. Intestinal Alkaline Phosphatase Attenuates Alcohol-Induced Hepatosteatosis in Mice. Dig. Dis. Sci. 2017, 62, 2021–2034.
- Rao, R.K. Acetaldehyde-induced barrier disruption and paracellular permeability in Caco-2 cell monolayer. Methods Mol. Biol. 2008, 447, 171–183.
- Uesugi, T.; Froh, M.; Arteel, G.E.; Bradford, B.U.; Thurman, R.G. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology 2001, 34, 101–108.
- Adachi, Y.; Moore, L.E.; Bradford, B.U.; Gao, W.; Thurman, R.G. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 1995, 108, 218–224.
- Elamin, E.E.; Masclee, A.A.; Dekker, J.; Jonkers, D.M. Ethanol metabolism and its effects on the intestinal epithelial barrier. Nutr. Rev. 2013, 71, 483–499.
- Nair, S.; Cope, K.; Risby, T.H.; Diehl, A.M. Obesity and female gender increase breath ethanol concentration: Potential implications for the pathogenesis of nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2001, 96, 1200–1204.
- Cope, K.; Risby, T.; Diehl, A.M. Increased gastrointestinal ethanol production in obese mice: Implications for fatty liver disease pathogenesis. Gastroenterology 2000, 119, 1340–1347.
- Zeisel, S.H.; da Costa, K.A. Choline: An essential nutrient for public health. Nutr. Rev. 2009, 67, 615–623.
- Han, J.; Dzierlenga, A.L.; Lu, Z.; Billheimer, D.D.; Torabzadeh, E.; Lake, A.D.; Li, H.; Novak, P.; Shipkova, P.; Aranibar, N.; et al. Metabolomic profiling distinction of human nonalcoholic fatty liver disease progression from a common rat model. Obesity 2017, 25, 1069–1076.
- Muraki, Y.; Makita, Y.; Yamasaki, M.; Amano, Y.; Matsuo, T. Elevation of liver endoplasmic reticulum stress in a modified choline-deficient l-amino acid-defined diet-fed non-alcoholic steatohepatitis mouse model. Biochem. Biophys. Res. Commun. 2017, 486, 632–638.
- Zeisel, S.H.; Wishnok, J.S.; Blusztajn, J.K. Formation of methylamines from ingested choline and lecithin. J. Pharmacol. Exp. Ther. 1983, 225, 320–324.
- Al-Waiz, M.; Mikov, M.; Mitchell, S.C.; Smith, R.L. The exogenous origin of trimethylamine in the mouse. Metabolism 1992, 41, 135–136.
- Lang, D.H.; Yeung, C.K.; Peter, R.M.; Ibarra, C.; Gasser, R.; Itagaki, K.; Philpot, R.M.; Rettie, A.E. Isoform specificity of trimethylamine N-oxygenation by human flavin-containing monooxygenase (FMO) and P450 enzymes: Selective catalysis by FMO3. Biochem. Pharmacol. 1998, 56, 1005–1012.
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584.
- Arslan, N. Obesity, fatty liver disease and intestinal microbiota. World J. Gastroenterol. 2014, 20, 16452–16463.
- Dumas, M.E.; Barton, R.H.; Toye, A.; Cloarec, O.; Blancher, C.; Rothwell, A.; Fearnside, J.; Tatoud, R.; Blanc, V.; Lindon, J.C.; et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12511–12516.
- Odamaki, T.; Kato, K.; Sugahara, H.; Hashikura, N.; Takahashi, S.; Xiao, J.Z.; Abe, F.; Osawa, R. Age-related changes in gut microbiota composition from newborn to centenarian: A cross-sectional study. BMC Microbiol. 2016, 16, 90.
- Francino, M.P. Antibiotics and the Human Gut Microbiome: Dysbioses and Accumulation of Resistances. Front. Microbiol. 2015, 6, 1543.
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Manneras-Holm, L.; Stahlman, M.; Olsson, L.M.; Serino, M.; Planas-Felix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858.
- Rogers, M.A.M.; Aronoff, D.M. The influence of non-steroidal anti-inflammatory drugs on the gut microbiome. Clin. Microbiol. Infect 2016, 22, 178.e1–e178.e9.
- Hill, D.A.; Siracusa, M.C.; Abt, M.C.; Kim, B.S.; Kobuley, D.; Kubo, M.; Kambayashi, T.; Larosa, D.F.; Renner, E.D.; Orange, J.S.; et al. Commensal bacteria-derived signals regulate basophil hematopoiesis and allergic inflammation. Nat. Med. 2012, 18, 538–546.
- Deplancke, B.; Gaskins, H.R. Microbial modulation of innate defense: Goblet cells and the intestinal mucus layer. Am. J. Clin. Nutr. 2001, 73, 1131S–1141S.
- Gordon, H.A.; Pesti, L. The gnotobiotic animal as a tool in the study of host microbial relationships. Bacteriol. Rev. 1971, 35, 390–429.
- Smith, K.; McCoy, K.D.; Macpherson, A.J. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin. Immunol. 2007, 19, 59–69.
- Lecuyer, E.; Rakotobe, S.; Lengline-Garnier, H.; Lebreton, C.; Picard, M.; Juste, C.; Fritzen, R.; Eberl, G.; McCoy, K.D.; Macpherson, A.J.; et al. Segmented filamentous bacterium uses secondary and tertiary lymphoid tissues to induce gut IgA and specific T helper 17 cell responses. Immunity 2014, 40, 608–620.
- Weinstein, P.D.; Cebra, J.J. The preference for switching to IgA expression by Peyer’s patch germinal center B cells is likely due to the intrinsic influence of their microenvironment. J. Immunol. 1991, 147, 4126–4135.
- Bouskra, D.; Brezillon, C.; Berard, M.; Werts, C.; Varona, R.; Boneca, I.G.; Eberl, G. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 2008, 456, 507–510.
- Macpherson, A.J.; Martinic, M.M.; Harris, N. The functions of mucosal T cells in containing the indigenous commensal flora of the intestine. Cell Mol. Life Sci. 2002, 59, 2088–2096.
- Macpherson, A.J.; Hunziker, L.; McCoy, K.; Lamarre, A. IgA responses in the intestinal mucosa against pathogenic and non-pathogenic microorganisms. Microbes Infect. 2001, 3, 1021–1035.
- Hall, J.A.; Bouladoux, N.; Sun, C.M.; Wohlfert, E.A.; Blank, R.B.; Zhu, Q.; Grigg, M.E.; Berzofsky, J.A.; Belkaid, Y. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity 2008, 29, 637–649.
- Uzbay, T. Germ-free animal experiments in the gut microbiota studies. Curr. Opin. Pharmacol. 2019, 49, 6–10.
- Hernandez-Chirlaque, C.; Aranda, C.J.; Ocon, B.; Capitan-Canadas, F.; Ortega-Gonzalez, M.; Carrero, J.J.; Suarez, M.D.; Zarzuelo, A.; Sanchez de Medina, F.; Martinez-Augustin, O. Germ-free and Antibiotic-treated Mice are Highly Susceptible to Epithelial Injury in DSS Colitis. J. Crohns Colitis 2016, 10, 1324–1335.
- Al-Asmakh, M.; Zadjali, F. Use of Germ-Free Animal Models in Microbiota-Related Research. J. Microbiol. Biotechnol. 2015, 25, 1583–1588.
- Tada, T.; Yamamura, S.; Kuwano, Y.; Abo, T. Level of myelopoiesis in the bone marrow is influenced by intestinal flora. Cell Immunol. 1996, 173, 155–161.
- Staber, F.G.; Tarcsay, L.; Dukor, P. Modulations of myelopoiesis in vivo by chemically pure preparations of cell wall components from gram-negative bacteria: Effects at different stages. Infect Immun. 1978, 20, 40–49.
- Khosravi, A.; Yanez, A.; Price, J.G.; Chow, A.; Merad, M.; Goodridge, H.S.; Mazmanian, S.K. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe 2014, 15, 374–381.
- Lee, Y.K.; Menezes, J.S.; Umesaki, Y.; Mazmanian, S.K. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4615–4622.
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The microbiome and innate immunity. Nature 2016, 535, 65–74.
- Vijay-Kumar, M.; Aitken, J.D.; Carvalho, F.A.; Cullender, T.C.; Mwangi, S.; Srinivasan, S.; Sitaraman, S.V.; Knight, R.; Ley, R.E.; Gewirtz, A.T. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010, 328, 228–231.
- Ramanan, D.; Tang, M.S.; Bowcutt, R.; Loke, P.; Cadwell, K. Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity 2014, 41, 311–324.
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734.
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalova, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443.
- Chudnovskiy, A.; Mortha, A.; Kana, V.; Kennard, A.; Ramirez, J.D.; Rahman, A.; Remark, R.; Mogno, I.; Ng, R.; Gnjatic, S.; et al. Host-Protozoan Interactions Protect from Mucosal Infections through Activation of the Inflammasome. Cell 2016, 167, 444–456 e414.
- Gury-BenAri, M.; Thaiss, C.A.; Serafini, N.; Winter, D.R.; Giladi, A.; Lara-Astiaso, D.; Levy, M.; Salame, T.M.; Weiner, A.; David, E.; et al. The Spectrum and Regulatory Landscape of Intestinal Innate Lymphoid Cells Are Shaped by the Microbiome. Cell 2016, 166, 1231–1246.e1213.
- Li, F.; Hao, X.; Chen, Y.; Bai, L.; Gao, X.; Lian, Z.; Wei, H.; Sun, R.; Tian, Z. The microbiota maintain homeostasis of liver-resident gammadeltaT-17 cells in a lipid antigen/CD1d-dependent manner. Nat. Commun. 2017, 7, 13839.
- Honda, K.; Littman, D.R. The microbiota in adaptive immune homeostasis and disease. Nature 2016, 535, 75–84.
- Peterson, D.A.; McNulty, N.P.; Guruge, J.L.; Gordon, J.I. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe 2007, 2, 328–339.
- Kunisawa, J.; Kiyono, H. Alcaligenes is Commensal Bacteria Habituating in the Gut-Associated Lymphoid Tissue for the Regulation of Intestinal IgA Responses. Front Immunol. 2012, 3, 65.
- Bunker, J.J.; Flynn, T.M.; Koval, J.C.; Shaw, D.G.; Meisel, M.; McDonald, B.D.; Ishizuka, I.E.; Dent, A.L.; Wilson, P.C.; Jabri, B.; et al. Innate and Adaptive Humoral Responses Coat Distinct Commensal Bacteria with Immunoglobulin A. Immunity 2015, 43, 541–553.
- Pabst, O. New concepts in the generation and functions of IgA. Nat. Rev. Immunol. 2012, 12, 821–832.
- Bemark, M.; Boysen, P.; Lycke, N.Y. Induction of gut IgA production through T cell-dependent and T cell-independent pathways. Ann. N. Y. Acad. Sci. 2012, 1247, 97–116.
- Macpherson, A.J.; Gatto, D.; Sainsbury, E.; Harriman, G.R.; Hengartner, H.; Zinkernagel, R.M. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science 2000, 288, 2222–2226.
- Moon, C.; Baldridge, M.T.; Wallace, M.A.; Burnham, C.A.D.; Virgin, H.W.; Stappenbeck, T.S. Vertically transmitted faecal IgA levels determine extra-chromosomal phenotypic variation. Nature 2015, 521, 90–93.
- Fransen, F.; Zagato, E.; Mazzini, E.; Fosso, B.; Manzari, C.; El Aidy, S.; Chiavelli, A.; D’Erchia, A.M.; Sethi, M.K.; Pabst, O.; et al. BALB/c and C57BL/6 Mice Differ in Polyreactive IgA Abundance, which Impacts the Generation of Antigen-Specific IgA and Microbiota Diversity. Immunity 2015, 43, 527–540.
- Viladomiu, M.; Kivolowitz, C.; Abdulhamid, A.; Dogan, B.; Victorio, D.; Castellanos, J.G.; Woo, V.; Teng, F.; Tran, N.L.; Sczesnak, A.; et al. IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote TH17-dependent inflammation. Sci. Transl. Med. 2017, 9.
- Atarashi, K.; Tanoue, T.; Ando, M.; Kamada, N.; Nagano, Y.; Narushima, S.; Suda, W.; Imaoka, A.; Setoyama, H.; Nagamori, T.; et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 2015, 163, 367–380.
- Britton, G.J.; Contijoch, E.J.; Mogno, I.; Vennaro, O.H.; Llewellyn, S.R.; Ng, R.; Li, Z.; Mortha, A.; Merad, M.; Das, A.; et al. Microbiotas from Humans with Inflammatory Bowel Disease Alter the Balance of Gut Th17 and RORgammat(+) Regulatory T Cells and Exacerbate Colitis in Mice. Immunity 2019, 50, 212–224.e214.
- Tanoue, T.; Atarashi, K.; Honda, K. Development and maintenance of intestinal regulatory T cells. Nat. Rev. Immunol. 2016, 16, 295–309.
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341.
- Round, J.L.; Mazmanian, S.K. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl. Acad. Sci. USA 2010, 107, 12204–12209.
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573.
- Park, J.; Kim, M.; Kang, S.G.; Jannasch, A.H.; Cooper, B.; Patterson, J.; Kim, C.H. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. 2015, 8, 80–93.
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772.
- Weiss, D.S. The great escape: Microbiotal LPS takes a toll on the liver. Cancer Prev. Res. 2012, 5, 1078–1080.
- Carpino, G.; Del Ben, M.; Pastori, D.; Carnevale, R.; Baratta, F.; Overi, D.; Francis, H.; Cardinale, V.; Onori, P.; Safarikia, S.; et al. Increased Liver Localization of Lipopolysaccharides in Human and Experimental NAFLD. Hepatology 2019.
- Szabo, G.; Velayudham, A.; Romics, L., Jr.; Mandrekar, P. Modulation of non-alcoholic steatohepatitis by pattern recognition receptors in mice: The role of toll-like receptors 2 and 4. Alcohol Clin. Exp. Res. 2005, 29, 140S–145S.
- Ramos, H.C.; Rumbo, M.; Sirard, J.C. Bacterial flagellins: Mediators of pathogenicity and host immune responses in mucosa. Trends Microbiol. 2004, 12, 509–517.
- Xiao, Y.; Liu, F.; Yang, J.; Zhong, M.; Zhang, E.; Li, Y.; Zhou, D.; Cao, Y.; Li, W.; Yu, J.; et al. Over-activation of TLR5 signaling by high-dose flagellin induces liver injury in mice. Cell Mol. Immunol. 2015, 12, 729–742.
- Weiss, E.; Kretschmer, D. Formyl-Peptide Receptors in Infection, Inflammation, and Cancer. Trends Immunol. 2018, 39, 815–829.
- Liu, M.; Chen, K.; Yoshimura, T.; Liu, Y.; Gong, W.; Wang, A.; Gao, J.L.; Murphy, P.M.; Wang, J.M. Formylpeptide receptors are critical for rapid neutrophil mobilization in host defense against Listeria monocytogenes. Sci. Rep. 2012, 2, 786.
- Pasare, C.; Medzhitov, R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 2003, 299, 1033–1036.
- Roh, Y.S.; Zhang, B.; Loomba, R.; Seki, E. TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G30–G41.
- Bergheim, I.; Weber, S.; Vos, M.; Kramer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992.
- Li, Z.; Yang, S.; Lin, H.; Huang, J.; Watkins, P.A.; Moser, A.B.; Desimone, C.; Song, X.Y.; Diehl, A.M. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003, 37, 343–350.
- Cani, P.D.; Neyrinck, A.M.; Fava, F.; Knauf, C.; Burcelin, R.G.; Tuohy, K.M.; Gibson, G.R.; Delzenne, N.M. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 2007, 50, 2374–2383.
- Wagnerberger, S.; Spruss, A.; Kanuri, G.; Stahl, C.; Schroder, M.; Vetter, W.; Bischoff, S.C.; Bergheim, I. Lactobacillus casei Shirota protects from fructose-induced liver steatosis: A mouse model. J. Nutr. Biochem. 2013, 24, 531–538.
- Esposito, E.; Iacono, A.; Bianco, G.; Autore, G.; Cuzzocrea, S.; Vajro, P.; Canani, R.B.; Calignano, A.; Raso, G.M.; Meli, R. Probiotics reduce the inflammatory response induced by a high-fat diet in the liver of young rats. J. Nutr. 2009, 139, 905–911.
- Daubioul, C.A.; Horsmans, Y.; Lambert, P.; Danse, E.; Delzenne, N.M. Effects of oligofructose on glucose and lipid metabolism in patients with nonalcoholic steatohepatitis: Results of a pilot study. Eur. J. Clin. Nutr. 2005, 59, 723–726.
- Jena, P.K.; Sheng, L.; Nagar, N.; Wu, C.; Barile, D.; Mills, D.A.; Wan, Y.Y. Synbiotics Bifidobacterium infantis and milk oligosaccharides are effective in reversing cancer-prone nonalcoholic steatohepatitis using western diet-fed FXR knockout mouse models. J. Nutr. Biochem. 2018, 57, 246–254.
- Dhiman, R.K.; Rana, B.; Agrawal, S.; Garg, A.; Chopra, M.; Thumburu, K.K.; Khattri, A.; Malhotra, S.; Duseja, A.; Chawla, Y.K. Probiotic VSL#3 reduces liver disease severity and hospitalization in patients with cirrhosis: A randomized, controlled trial. Gastroenterology 2014, 147, 1327–1337.e1323.
- Bajaj, J.S.; Kassam, Z.; Fagan, A.; Gavis, E.A.; Liu, E.; Cox, I.J.; Kheradman, R.; Heuman, D.; Wang, J.; Gurry, T.; et al. Fecal microbiota transplant from a rational stool donor improves hepatic encephalopathy: A randomized clinical trial. Hepatology 2017, 66, 1727–1738.
- Wang, W.W.; Zhang, Y.; Huang, X.B.; You, N.; Zheng, L.; Li, J. Fecal microbiota transplantation prevents hepatic encephalopathy in rats with carbon tetrachloride-induced acute hepatic dysfunction. World J. Gastroenterol. 2017, 23, 6983–6994.
- Vetizou, M.; Pitt, J.M.; Daillere, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084.
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089.