Baeyer–Villiger monooxygenases (BVMOs) are flavin-dependent oxidative enzymes capable of catalyzing the insertion of an oxygen atom between a carbonylic Csp2 and the Csp3 at the alpha position, therefore transforming linear and cyclic ketones into esters and lactones. These enzymes are dependent on nicotinamides (NAD(P)H) for the flavin reduction and subsequent reaction with molecular oxygen. BVMOs can be included in cascade reactions, coupled to other redox enzymes, such as alcohol dehydrogenases (ADHs) or ene-reductases (EREDs), so that the direct conversion of alcohols or α,β-unsaturated carbonylic compounds to the corresponding esters can be achieved.
- biocatalysis
- whole cells
- cascade reactions
- redox enzymes
- monooxygenases
- Baeyer–Villiger alcohol dehydrogenases
- ene-reductases
1. Introduction
The elucidation and rational understanding of the internal organization of the different biocatalytic reactions occurring inside biological cells, in which several enzymatic reactions proceed in a concatenated manner, is one of the basis of Systems Biology [1]. The development of this discipline has fostered the design of coupled systems of biocatalytic reactions, and the number of publications dealing with this topic has increased considerably during the last years [2][3][4][5][6][7][8][9][10][11]. In this area, oxidoreductases are enzymes frequently used in cascade reactions, as usually a pair of these enzymes are simultaneously applied for the required in situ recycling of the cofactors [12][13][14][15]. Inside this type of enzymes, Baeyer–Villiger monooxygenases (BVMOs) are undoubtedly one of the most attractive members of this family; these flavin-dependent oxidative enzymes [16][17][18][19][20] are capable of catalyzing the insertion of an oxygen atom between a carbonylic Csp2 and the Csp3 at the alpha position, therefore transforming linear and cyclic ketones into esters and lactones [20][21][22][23][24][25][26][27], as schematized in Scheme 1.
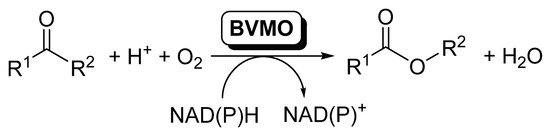
Scheme 1. BVMOs-catalyzed oxidation of ketones to furnish esters.
BVMOs require nicotinamides (NADPH) for the flavin reduction to FADH2, which reacts with molecular oxygen to form the reactive peroxyflavin responsible for the substrate oxidation. As NADPH is an expensive compound and its presence at high concentrations can inhibit the biocatalyst, it has to be recycled to obtain feasible biocatalytic procedures. In the last years, more than 100 BVMOs have been cloned and overexpressed. In many cases, the natural role of those BVMOs is not identified, while in others they seem to take part in the synthesis of secondary metabolites. Most of the BVMOs, the so-called type I BVMOs, can be included in the B subclass of flavin dependent monooxygenases [28], according to their protein sequence motifs, electron donor and type of oxygenation reaction. Thus, they are single-component enzymes possessing two α/β Rossmann-like domains for the FAD and NADPH binding, respectively, and they keep this last one bound during catalysis, while the substrate binds after the flavin-peroxide is formed [17]. BVMO catalysis started by NADPH binding and subsequent flavin reduction, after which the NADPH cofactor adopts a stable position. A stable peroxyflavin is the catalytically active specie, formed by the reaction of the reduced flavin with molecular oxygen [29]. Type II BVMOs are part of the class C of flavin dependent monooxygenases, which are two-component monooxygenases [30], but this type of BVMOs has been scarcely employed in biocatalysis due the requirement of these two components.
Most of the BVMOs are present on prokaryotes and some unicellular eukaryotic organisms such as filamentous fungi. Several bacterial BVMOs have been applied with biocatalytic purposes; even some fungal BVMOs have also been discovered and characterized in the last few years [31]. Both storage and operational stability of these biocatalysts is interesting for their biotechnological applications. For most of the BVMOs, which presents a certain stability, lyophilization in presence of different additives is a method of interest because it simplifies the enzyme transport and storage [32]. Normally, these biocatalysts perform their activity at mild reaction conditions (aqueous media at neutral pH and room temperature). In the last few years several examples of thermostable BVMOs (wild type and mutants), able to catalyze reactions at high temperatures, have been reported [33][34], with the aim of increasing the applicability of these valuable enzymes [35][36].
Baeyer–Villiger monooxygenases were first discovered in the 1960s, but they have not been widely applied with biocatalytic purposes for the preparation of high valuable compounds as esters, lactones and sulfoxides, among others. Advances in genome mining have allowed scientists to discover several BVMOs active on different types of compounds, but still nowadays, cyclohexanone monooxygenase (CHMO) from Acinetobacter calcoaceticus NCIMB 9871, discovered more than 40 years ago [37], is the BVMO with the highest applicability due to its large substrate profile and excellent selectivity. Nevertheless, CHMO presents some drawbacks as its thermal instability and its low stability to organic co-solvents [38]. Remarkably, most BVMOs display an excellent behavior when employed in multi-step cascade reactions coupled to other enzymes. In most of these reactions, lactones are obtained as final products. These cyclic esters of carboxylic acids, containing a 1-oxacycloalkan-2-one structure, are a class of secondary metabolites, thus presenting a wide range of biological activities as anti-inflammatory, antimicrobial or anticancer compounds [39]. Lactones can be also employed in cosmetics and perfume industry, in polymer chemistry, as agrochemicals or in food industry as flavoring agents. By this reason, several methodologies have been performed for their preparation. The use of biocatalysts for the synthesis of lactones presents some advantages regarding the classical methods, as only molecular oxygen is required as oxidant while working under mild (pH and temperature) reaction conditions, being achieved in general high regio- and/or enantioselectivites. The combination of two or more biocatalysts also allowed avoiding the isolation and purification of the reaction intermediates, thus increasing the atom economy of the processes. However, the use of BVMOs in these synthetic procedures can present some limitations, as low enzyme expression and stability, the NADPH-dependence, and substrate and product inhibition.
2. Multi-Step Reactions Including BVMO Activity Catalyzed by Whole Cells
Generally speaking, the use of whole cells makes the cascade easier compared to those employing isolated biocatalysts, as the recycling of the required cofactors is produced inside the cell metabolic machinery, so that it is not mandatory to implement an external recycling methodology. The most attractive methodology using whole cells implies the use of à la carte engineered cells, inside which different enzymatic activities are overexpressed in order to generate the desired multi-step procedure [40]. This methodology, which has been sometimes termed as Systems Biocatalysis [41], presents several advantages [40][42][43][44][45]: (i) as already mentioned, the intra-cellular medium provides the natural enzymatic environment and the cofactors regeneration machinery; (ii) it is relatively easy and economical to have available cells by cultivation without any additional downstream process; (iii) enzymes inside cell walls and membranes are somehow protected from extreme reaction conditions, and (iv) the different enzymes involved in the cascade are co-localized inside cells, so that their local concentration is increased, therefore reducing the diffusion of intermediates.
2.1. Multi-Step Reactions Including Alcohol Dehydrogenases/BVMO Activity Catalyzed by Whole Cells
Most of the examples of biocatalytic cascades employing BVMOs have been developed together with alcohol dehydrogenases (ADHs, EC 1.1.1.x), also called ketoreductases (KREDs). These enzymes are able to catalyze the reversible reduction of carbonyl compounds into the corresponding alcohols (Scheme 2), in general with high selectivity [46][47][48][49]. ADHs require the presence of nicotinamide cofactors for performing their activity, being required to employ effective regeneration systems if these enzymes are employed as isolated biocatalysts.
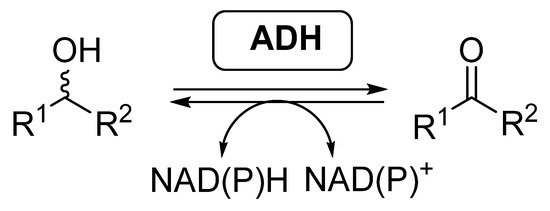
Scheme 2. Alcohol dehydrogenases catalyzed reversible reduction of carbonyl compounds and oxidation of alcohols.
Initial studies on linear cascades combining ADHs and BVMOs were performed at the beginning of the 1990s, using whole cells systems for the direct bio-oxidation of alcohols to the corresponding lactones. Thus, the starting alcohol was oxidized to the corresponding ketones by the ADH at the expense of NAD(P)+, whereas the obtained ketone was further oxidized in presence of the BVMO to the desired lactone, employing NAD(P)H which was converted again into NAD(P)+. A pioneer example is the oxidation of alcohol 1, shown in Scheme 3, employing fractured cells of Acinetobacter calcoacetitus NCIMB to the corresponding lactone 3, described in 1991 [50]. At short reaction times, 1 was converted into ketone 2 in a great extent (80% after 6 h). Longer reaction time led to an increase in the lactone production, reaching a maximum value of 40% after 48 h, whereas concentration of 2 started to decrease after 10 h. A similar pattern was observed in the bio-oxidation of both endo- and exo-bicyclo [2.2.1]heptan-2-ols. Alcohol concentration decreased as the ketone became formed and lactone concentration started to increase during the course of the biocatalytic process. Fractured cells of A. calcoaceticus were also employed in the bio-oxidation of 6-endo-bicyclo [3.2.0]hept-2-en-6-ol 4 to obtain regioisomeric lactones 5 and 6 with complete conversion after 1 h, as shown in Scheme 3. A 15% of a by-product, which authors assumed that was exo-4, was observed in the crude mixture.
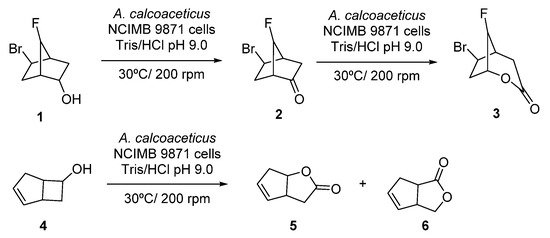
Scheme 3. Acinetobacter calcoaceticus cells catalyzed biotransformations of alcohols into lactones.
The combination of BVMOs and ADHs has been further developed for the preparation of ε-caprolactone (ECL, 9, Scheme 4). This compound is a valuable material with several applications in the field of colorants, adhesives and coating materials, as ECL can be easily polymerized yielding biodegradable thermoplastics and elastomeric polymers [51][52]. By these reasons, several approaches have been studied for its preparation under sustainable reaction conditions. BVMOs and ADHs have been employed well as whole cells or well as isolated enzymes for the synthesis of ECL starting from cyclohexanol 7, in a process in which this alcohol is oxidized by the ADH to cyclohexanone 8, which is converted into ECL in presence of the BVMO, with an in situ NAD(P)H cofactor regeneration. When employing this approach, the stability of the BVMO is critical for the process, as this biocatalyst is very sensitive to the cyclohexanol and ECL concentration in the reaction medium. For this reason, several attempts have been made in order to overcome this bottleneck.
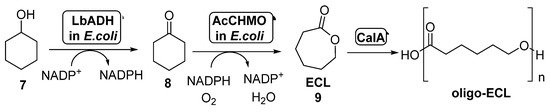
Scheme 4. Preparation of ECL 9 and its oligomers through a biocatalytic procedure employing whole cells of Lactobacillus brevis ADH and Acinetobacter calcoaceticus CHMO.
Thus, the bienzymatic system employing whole cells of ADH-BVMO for the preparation of ECL can be coupled with the lipase-catalyzed hydrolysis of this compound in order to obtain oligo-caprolactone. This approach was performed for the first time in 2015 [53]. In order to overcome the BVMO deactivation caused by the presence of ECL in the reaction medium, this product subjected to in situ ring-opening oligomerization catalyzed by CALA (lipase from Candida antarctica, Scheme 4). The formed oligo-ECLs were easily removed from the reaction by extraction or precipitation. CALA is able to catalyze the formation of the polymers even in presence of high amounts of water, not showing any hydrolytic activity on ECL at high substrate concentrations (1.0 M). After a few hours in presence of CALA, the lactone was converted into oligomers with a maximum molecular weight of 1200 g/mol, which can be transformed into high-molecular weight polymer. Recombinant E. coli cells of Lactobacillus brevis ADH (LbADH) and the stable mutant C376L/M400I of Acinetobacter calcoaceticus cyclohexanone monooxygenase (AcCHMO), at 100 g wet cells weight/mL concentration, were combined with lipase CALA (10 mg/mL lyophilized) in a one-pot process at different concentrations of cyclohexanol. A decrease in the ECL concentration in these reactions was detected when compared to those in absence of the lipase, thus indicating the beneficiary effect of the conversion of the ECL into oligo-ECL. When both the ADH and the BVMO were employed as separate cells, results were better than expressing both biocatalysts into the same cell-system. LbADH/AcCHMO ratio was optimized 1:10 in order to achieve the highest conversion. Addition of acetone and glucose as co-substrates was also positive for a faster regeneration of the endogenous NADPH in the cell systems. The optimized cascade was performed at preparative scale and, after 48 h, a complete conversion was achieved when starting from 200 mM of 7, yielding a 75% of oligo-ECL and a 25% of ECL. Higher substrate concentrations, 300 mM and 500 mM, afforded lower conversions, 74% and 43% respectively.
In a further development of this trienzymatic system, the same authors described the preparation of methyl-substituted ECL derivatives in a one-pot two-step process [54], as shown in Scheme 5.
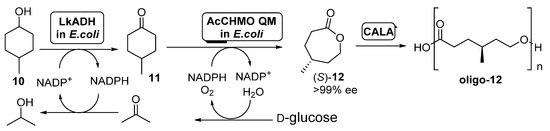
Scheme 5. Preparation of (S)-4-methylcaprolactone (S)-12 and its oligomers through a biocatalytic procedure employing whole cells of Lactobacillus kefir ADH and Acinetobacter calcoaceticus CHMO mutant QM.
A diastereomeric mixture of 4-methylcyclohexanol (10) was employed as starting substrate; under those conditions, AcCHMO was able to convert 4-methylcyclohexanone 11 into the enantiopure lactone (S)-12 with high yield and complete selectivity. Initial experiments were performed with E. coli cells expressing Lactobacillus kefir ADH (LkADH) and wild type AcCHMO in 1:10 ratio (100 g wet cells weight/mL), but the conversions at all concentrations tested (5–20 mM) were low, achieving only a 34% of the enantiopure lactone as highest value when working at 5 mM. Poor conversion values were caused by the low conversion in the ADH catalyzed oxidation, due to the inhibition of this biocatalyst by the ketone 11 formed. In order to improve this process, a faster second oxidation process, with the aim of the fast removing of 11 from the reaction medium was envisioned. Thus, a more stable mutant of AcCHMO was employed (C376L/M400I/T415C/A463C, the so-called AcCHMO QM) and the ratio of the E. coli cells for both biocatalysts was optimized to 1:1. The supply of pure oxygen to the reaction also resulted in a higher conversion, close to 90% after 24 h at 25 °C. The addition of CALA (10 mg/mL) to the enzymatic system led to a further decrease in (S)-12 concentration, as it was being hydrolyzed into the oligo-(S)-lactone.
The production of ECL starting from cyclohexanol employing E. coli cells of ADH and AcCHMO has been studied by using a computational approach in 2017 [55], applying a kinetic model for the study this cascade reaction in both batch and fed-batch synthesis. To this purpose, a fed-batch synthesis was developed in order to circumvent the CHMO inhibition caused by cyclohexanol, which hampered the use of this substrate at concentrations higher than 60 mM when employed in batch. Thus, 7 was added to the reaction medium at different ratios, from 14.2 to 19.0 μmol/min; feed rates higher than 17.5 μmol/min led to the accumulation of 7, therefore reducing the rate formation of ECL due to CHMO inhibition. The amount of ECL in the reaction medium was reduced by hydrolyzing this compound to 6-hydroxyhexanoic acid (13, Scheme 6) in a process catalyzed by Candida antarctica lipase B (CALB). After 6 h, 162 mM of 13 was obtained with a feed rate of 19 μmol/min. The addition of CALB maintained a low ECL concentration in the reaction medium (lower than 10 mM).
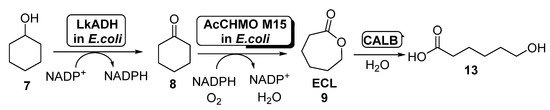
Scheme 6. Preparation of ECL 9 and its oligomers through a biocatalytic procedure employing whole cells of Lactobacillus brevis ADH and Acinetobacter calcoaceticus CHMO M15.
The production of ECL has been optimized by performing the co-expression of the BVMO and the ADH in E. coli, employing a DuetTM vector [56], in order to obtain a higher efficiency for the enzymatic cascade. The pRSFDuet plasmid form Novagen was selected as dual expression vector, cloning the AcCHMO QM gene into the first multiple cloning site and the LkADH gene into the second multiple cloning site. With the aim of balancing the expression levels of the cascade enzymes, the ADH gene was subjected to engineering of the ribosome binding site (RBS), employing the native RBS sequence as well as two point mutations. In order to compare the results, both types of enzymes were expressed separately into E. coli cells, performing the biotransformations with an optimized ratio of CHMO: ADH cells 5:1 (100 g wet cells weight/mL) and in presence of glucose and acetone and co-substrates, as higher conversions were obtained than in absence of these compounds. This result can be explained by the difficult diffusion of the NADPH formed in the E. coli cells of the LkADH to the AcCHMO QM cells. After 16 h, it was possible to achieve a 90% conversion from 20 mM of cyclohexanol employing wild type RBS, while much lower conversions were measured with the other two RBS mutations, indicating that a decreased LkADH expression is required to obtain a similar enzyme ratio for optimal conversion. When the same process was carried out with the E. coli cells containing the double gene, a complete conversion was obtained after 16 h for all the three RBS preparations in the absence of co-substrates. In presence of glucose and acetone, complete conversion was also observed for the CHMO co-expressed with one of the RBS mutations of the ADH after 16 h, whereas after 2 h, a 54% conversion was reached. This system was the most efficient for the co-expression of both biocatalysts, suggesting that the ratios of expressed AcCHMO QM and LkADH were similar in this system. When higher substrate concentrations were employed, lower conversion values were achieved, mainly debt to the inhibitory effect of both 7 and 9 on AcCHMO.
Very recently, the conversion of cyclohexanol to ECL by combining LkADH with two stable mutants of CHMO has been studied in order to obtain a suitable process at 200 mM scale. Different parameters that affect the process were analyzed and optimized in order to achieve the most productive process for the preparation of the final lactone [57]. The reaction was carried out in a stirred-tank reactor with maintained temperature and pH, bubbling a mixture of oxygen with synthetic air and pumping cyclohexanol at a constant rate proportional to the whole cell cascade activity. As both 7 and 8 present a high vapor pressure, they can be stripped out of the system by the off gas. The key component of the complete system, as previously stated, is CHMO, due to its low stability and the requirement of a proper oxygen supply. This last parameter was optimized by performing a bubble aeration technique. BVMO stability was improved by assessing two CHMO mutants; thus, AcCHMO QM showed a 40% higher long-term stability, whereas AcCHMO M15, containing eight mutations, has a higher oxidative stability combined with an improved thermostability. These two variants were compared employing E. coli cells co-expressing ADH and the CHMO at pH 7.5 and 30 °C, with 200 mM cyclohexanol fed at 4.5 mM/h. When using AcCHMO QM, the formation of ECL achieved a value of 45 mM after 13 h, but at longer reaction times this concentration decreased to 39 mM, probably debt to the ECL autohydrolysis to yield 13. The same experiment was performed with AcCHMO M15, reaching a maximum concentration of ECL after 18 h (79 mM). Longer reactions times resulted in substrate accumulation in the reactor with no further formation of the desired product. AcCHMO M15 showed longer activity, but it was not possible to achieve complete conversion. The stability of both CHMO mutants under the process conditions was analyzed; when employing AcCHMO QM, the biocatalyst retained its complete activity for one hour and it decreased to 85% after 8 h. After this reaction time, there was an important drop in the biocatalyst activity. On the other hand, AcCHMO M15 maintained its activity (98% of the initial one) even after 18 h. Thus, when working with AcCHMO QM, fresh cells were added after 8 h, whereas no extra addition was required for AcCHMO M15. Further experiments showed that an increase in the cells loading in the reactor led to a decrease in the volumetric mass transfer, thus resulting in a lower efficiency. The complete optimization of the biocatalytic process allowed scientists to achieve conversions higher than 98% for both CHMO mutants, being possible to obtain a higher final product concentration (21.1 g/L), space time yield (1.1 g/L h) and isolated ECL amount (9.1 g) employing AcCHMO M15, due to its higher operational stability. These values represent a 100% increase regarding the non-optimized processes, indicating the development of a more efficient process for the preparation of ECL.
Fusion proteins have been initially employed to enhance the soluble expression of proteins or to improve the enzyme purification, but in the last years some examples have appeared in which these systems have been used with synthetic purposes [58]. In 2015, the preparation of an enzyme fusion formed by the ADH from Micrococcus luteus NCTC2665 and the BVMO from Pseudomonas putida KT2440 (3.0 g dry cells/L), able to catalyze the double oxidation of long-chain unsaturated secondary alcohols (14a–c) to the corresponding esters (16a–c) through the corresponding ketones 15a–c, was reported [59], as shown in Scheme 7. These products can be hydrolyzed into valuable ω-hydroxycarboxylic acids and n-alkanoic acids, valuable compounds. The design of the fusion enzyme showed the highest expression level when using a glycine-rich linker, formed by 12 aminoacids, between the two biocatalysts. When the bio-oxidations were carried out with the fused biocatalyst expressed in E. coli, higher conversions were achieved for all the substrates when compared with the independent ADH and BVMO expressed in E. coli. Thus, esters 16a–c can be recovered with conversions higher than 75%, with activities higher than 22 µmol/g dry cells min. This positive effect of the fusion enzyme in the cascade activity can be debt to a higher functional expression of the BVMO and/or to a better mass transport efficiency, due to the presence of both active centers at close positions.
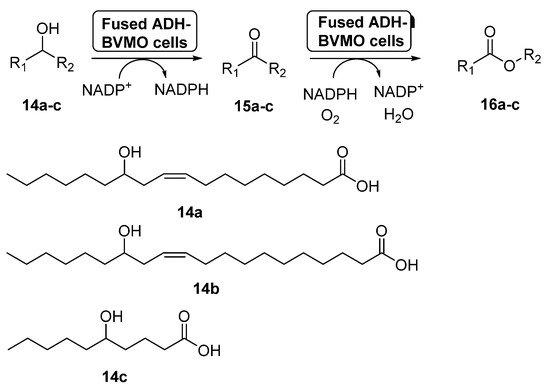
Scheme 7. Preparation of ECL 9 and its oligomers through a biocatalytic procedure employing whole cells of Lactobacillus brevis ADH and Acinetobacter calcoaceticus CHMO M15.
2.2. Multi-Step Reactions Including ERED/BVMO Activity Catalyzed by Whole Cells
Ene-reductases (EREDs) from the old yellow enzyme (OYE) family are flavin-dependent enzymes that catalyze the chemo- and stereo-selective asymmetric reduction of electronically activated carbon–carbon double bonds [25][27][60][61][62], as depicted in Scheme 8.
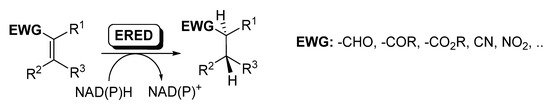
Scheme 8. Ene-reductases for the reduction of C=C.
Several examples can be found in literature illustrating the use of whole cells-catalyzed cascades coupling BVMO and ERED activities. For instance, Silva et al. [63] reported the preparation of ECL 9 starting from either cyclohexenone 17, cyclohexanone 8 or cyclohexanol 7, using whole cells of Brazilian Geotrichum candidum CCT 1205 (Scheme 9).
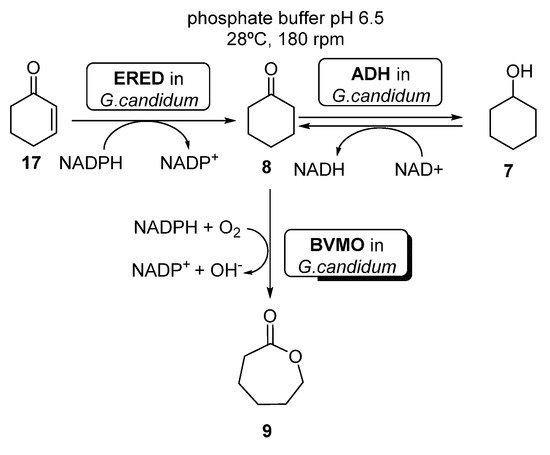
Scheme 9. Preparation of ε-caprolactone 9, using whole cells of Geotrichum candidum CCT 1205.
When using cyclohexanone 8 as substrate, these authors reported quantitative yields of the desired ε-caprolactone 9 after only 5 h, using a higher amount (3.0 g versus 1.0 g) of whole cells (otherwise, the cascade accumulated 7 and 8). Using 7 or 8 as substrates, the reaction was even faster (4 h) than using only 10 g of cells. These results are better than those previously reported by Mihovilovic et al. [64], leading to 52% of 9 in 48 h, using BVMO expressed in E. coli. Recently, Silva et al. [65] have shown that it is possible to immobilize the whole cells from Geotrichum candidum CCT 1205 in modified silica (n SiO2–Cl, SiO2–NH2 and SiO2–SH supports), without altering their catalytic performance and also allowing the stabilization and reuse of the cells.
Although the previous example illustrates the use of non-engineered cells, it is becoming more usual the use of genetically modified cells. Hence, Liu and Li described an enantioselective reduction−oxidation−hydrolysis cascade catalyzed by engineered whole cells for the synthesis of (R)-2-alkyl-δ-lactones 22–23 (useful flavor and fragrance materials) starting from the corresponding 2-alkylidenecyclopentanones 18–19 (Scheme 10) [66].
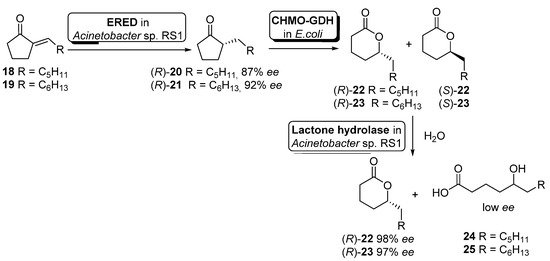
Scheme 10. Preparation of enantiopure (R)-2-alkyl-δ-lactones by using a whole-cell-catalyzed cascade involving BVMO activity.
In this example, the cascade catalysis was started with 50 mL of cell suspension of Acinetobacter sp. RS1 in Tris buffer (cell density 12 g cdw/L) containing 40 mg of alkylidene ketones 18 or 19 and 20 mg/mL glucose. After 3 h of reaction to allow the ERED-mediated accumulation of alkyl ketones 20 and 21 (R-configuration), 150 mL of Tris buffer containing engineered cells of E. coli (CHMO−GDH) (10 g cdw/L) was added to start the Baeyer–Villiger oxidation. After simultaneous oxidation and hydrolysis for 1.5 h, extraction with ethyl acetate and purification by flash chromatography, compounds (R)-22 (56% yield, 98% ee) or (R)-23 (41% yield, 97% ee) could be obtained. The presence of a hydrolytic activity inside Acinetobacter sp. RS1 cells was responsible for the hydrolysis of the intermediate (S)-22 or (S)-23 leading to δ-hydroxyacids 24 or 25, obtained with low enantioselectivity (E = 8–11).
The different enantiomers of carvolactones (7-methyl-4-(prop-1-en-2-yl)oxepan-2-ones) are gaining importance as monomers for the preparation of polymeric thermoplastic elastomers (shape-memory polymers) and pressure-sensitive adhesive components [67][68][69]. These carvolactones can be obtained starting from biogenic carvones after a C=C reduction and a Baeyer–Villiger oxidation. To this purpose, Iqbal et al. [70] have reported the concurrent redox cascade to transform (-) carvone and (+)-carvone [(R) and (S)-26] into the correspondent lactones, as depicted in Scheme 11.
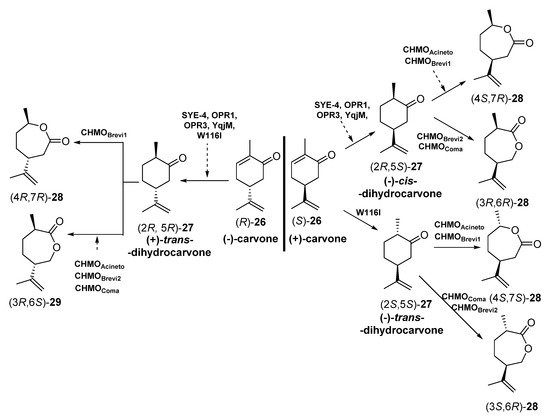
Scheme 11. Preparation of carvolactone stereoisomers 28 and 29 by a concurrent redox cascade.
Crude cell extracts (5.0 mg in 200 µL) from E. coli BL21 (DE3) containing the corresponding EREDs or BVMOs were used. Initially, each biotransformation step was carried out independently, starting with bioreductions of enantiomerically pure carvones (R) or (S)-26 and subsequent bio-oxidation with different BVMOs to furnish different stereoisomers of carvolactones 28 and 29. Finally, the reduction-oxidation steps were performed in a concurrent one-pot cascade. Both for the individual steps and the cascade, NADP+, glucose -6-phosphate and glucose -6-phosphate dehydrogenase were used for cofactor recycling. The EREDs that expressed E. coli BL21 (DE3) were SYE-4 (from Shewanella oneidensis [71]), OPR1 and OPR3 (12-oxophytodienoate reductase from Lycopersicon esculentum, tomato [72]), YqjM (from Bacillus subtilis [73]) and the variant W116I (replacement of tryptophan-116 by isoleucine in the OYE from Saccharomyces pastorianus [74]). On the other hand, the BVMOs were either cyclohexanone monooxygenases (CHMOs: AcCHMO [75], CHMOBrevi1 from Brevibacterium sp. [76] or cyclopentanone monooxygenases (CPMOs-type enzymes: CHMOBrevi2 from Brevibacterium sp. [76] and CPMOComa from Comamonas sp. [77]). Regarding the reaction stereochemistry of the bioreduction, as depicted in Scheme 5, when starting from (-)-carvone (R)-26 all the EREDs catalyzed the trans C=C bioreduction to afford (+)-trans-dihydrocarvone (2R,5R)-27. Opposite, if (+)-carvone (S)-26 was the initial substrate, most of the EREDs catalyzed the cis C=C bioreduction to afford (+)-cis-dihydrocarvone (2R,5S)-27, while the trans bioreduction, leading to (-)-trans-dihydrocarvone (2S,5S)-27, was observed only when employing W116I. Analyzing the regioselectivity of the BVMOs, depending on the absolute configuration of 27, some of the enzymes were leading to the “normal” lactones 28 (classical migration of the Criegge intermediate leading to the oxygen insertion in the most substituted alpha carbon [78]) or the “abnormal” ones 29, in good yields and enantiopurity.
In another example, Oberleitner et al. [79] reported the preparation of carvolactone (4S,7R)-28 starting from (R)-limonene ((R)-30) directly extracted from orange peel by means of a biocatalytic cascade shown in Scheme 12.
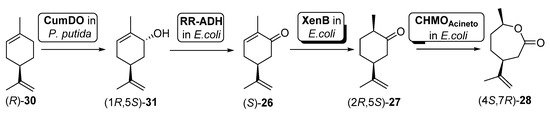
Scheme 12. Preparation of carvolactone (4S,7R)-28, using a whole cells-catalyzed cascade involving a BVMO.
To this purpose, two different types of cells were used: (i) an engineered Pseudomona putida S12 expressing cumene dioxygenase (CumDO) and (ii) E. coli BL21 (DE3) cells possessing different oxidative activities. These last ones were previously reported as being capable of catalyzing the conversion from carveol (1R,5S)-31 into (4S,7R)-28 [80], as they contain the expression of an alcohol dehydrogenase (RR-ADH from Rhodococcus ruber), an ERED (XenB from Pseudomonas sp.) and a BVMO (AcCHMO). Through the mixed culture approach, combining the two bacterial strains (different concentrations tested) in one pot, 47% of (4S,7R)-28 was produced after 20 h. They used a sequential approach, where hydroxylation of (R)-30 by CumDO was performed first, and E. coli BL21(DE3) resting cells were added to the reaction vessel after 10 h, observing full conversion to (4S,7R)-28 in 20 h. Then, these authors explored the direct use of waste product orange peel (biomass loading of about 3% (w/v), obtaining 3.2 mg of (4S,7R)-28 per g orange peel. Anyhow, by lowering the orange peel amount to 1.5% (w/v) and using the mixed-culture sequential combination, it was possible to furnish 6.3 mg of carvolactone per g orange peel (29% of (4S,7R)-28 from (R)-30 over four biocatalytic steps, 73% per step), only relying on orange peel as the substrate reservoir in aqueous buffer without any additives.
Recently, Avalos et al. [81] have described a similar process, using engineered E. coli cells, already designed to follow the Mentha spicata route leading to (-)-carvone (R)-26, in which the addition of a ERED and a BVMO (10 µM) activity finally furnished (4R,7R)-28, also known as (+)-dihydrocarvide (+)-DHCD, as shown in Scheme 13.
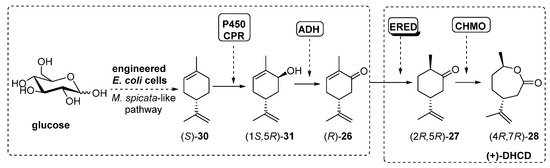
Scheme 13. Preparation of (+)-DHCD, using engineered whole-cells-catalyzed cascade.
The initial step into the M. spicata biosynthesis of (-)-carvone (R)-26 is the hydroxylation of (S)-limonene (S)-30 to furnish carveol (1S,5R)-31 catalyzed by limonene-6-hydroxylase (L6H, a P450 enzyme) coupled to SmCPR, a cytochrome P450 reductase from Salvia miltiorrhiza. Based on this system, authors constructed the rest of the enzymatic system by adding genes from ADH (RR-ADH was the best option), ERED (PETNR, pentaerythriol tetranitrate reductase from Enterobacter cloacae PB2 [82][83]) and BVMO (CHMO3M, a triple mutant from Rhodococcus sp. Phi1 CHMO [84]). Under optimal conditions, the complete biosynthesis of (+)-DHCD from glucose in E. coli was reported at 6.6 mg/L.
This entry is adapted from the peer-reviewed paper 10.3390/catal11050605
References
- Kitano, H. Systems biology: A brief overview. Science 2002, 295, 1662–1664.
- García-Junceda, E. Multi-Step Enzyme Catalysis: Biotransformations and Chemoenzymatic Synthesis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008.
- García-Junceda, E.; Lavandera, I.; Rother, D.; Schrittwieser, J.H. (Chemo)enzymatic cascades-Nature’s synthetic strategy transferred to the laboratory. J. Mol. Catal. B Enzym. 2015, 114, 1–6.
- Oroz-Guinea, I.; Fernández-Lucas, J.; Hormigo, D.; García-Junceda, E. Designed Enzymatic Cascades. In Science of Synthesis: Biocatalysis in Organic Synthesis 3; Faber, K., Fessner, W.D., Turner, N.J., Eds.; Georg Thieme Verlag: Stuttgart, Germany, 2015; Volume 3.
- Schrittwieser, J.H.; Velikogne, S.; Hall, M.; Kroutil, W. Artificial Biocatalytic Linear Cascades for Preparation of Organic Molecules. Chem. Rev. 2018, 118, 270–348.
- Schmidt, S.; Castiglione, K.; Kourist, R. Overcoming the Incompatibility Challenge in Chemoenzymatic and Multi-Catalytic Cascade Reactions. Chem. Eur. J. 2018, 24, 1755–1768.
- Schrittwieser, J.H.; Velikogne, S.; Kroutil, W. Artificial Biocatalytic Cascades to Alcohols and Amines. In Modern Biocatalysis: Advances Towards Synthetic Biological Systems; Williams, G., Hall, M., Eds.; Royal Soc Chemistry: Cambridge, UK, 2018; Volume 32, pp. 387–438.
- Walsh, C.T.; Moore, B.S. Enzymatic Cascade Reactions in Biosynthesis. Angew. Chem. Int. Ed. 2019, 58, 6846–6879.
- Kara, S.; Rudroff, F. (Eds.) Enzyme Cascade Design and Modelling; Springer International Publishing: Cham, Switzerland, 2021.
- McIntosh, J.A.; Owens, A.E. Enzyme engineering for biosynthetic cascades. Curr. Opin. Green Sustain. Chem. 2021, 29, 100448.
- Nazor, J.; Liu, J.; Huisman, G. Enzyme evolution for industrial biocatalytic cascades. Curr. Opin. Biotechnol. 2021, 69, 182–190.
- Uppada, V.; Bhaduri, S.; Noronha, S.B. Cofactor regeneration—An important aspect of biocatalysis. Curr. Sci. 2014, 106, 946–957.
- Berenguer-Murcia, A.; Fernandez-Lafuente, R. New Trends in the Recycling of NAD(P)H for the Design of Sustainable Asymmetric Reductions Catalyzed by Dehydrogenases. Curr. Org. Chem. 2010, 14, 1000–1021.
- Truppo, M.D. 7.4 Cofactor Recycling for Enzyme Catalyzed Processes. In Comprehensive Chirality; Elsevier Ltd.: Amsterdam, The Netherlands, 2012; Volume 7, pp. 46–70.
- Mordhorst, S.; Andexer, J.N. Round, round we go-strategies for enzymatic cofactor regeneration. Nat. Prod. Rep. 2020, 37, 1316–1333.
- Reis, R.A.G.; Li, H.; Johnson, M.; Sobrado, P. New frontiers in flavin-dependent monooxygenases. Arch. Biochem. Biophys. 2021, 699, 14.
- Toplak, M.; Matthews, A.; Teufel, R. The devil is in the details: The chemical basis and mechanistic versatility of flavoprotein monooxygenases. Arch. Biochem. Biophys. 2021, 698, 14.
- Dockrey, S.A.B.; Narayan, A.R.H. Flavin-dependent biocatalysts in synthesis. Tetrahedron 2019, 75, 1115–1121.
- Hall, M. Flavoenzymes for biocatalysis. In Enzymes; Chaiyen, P., Tamanoi, F., Tamanoi, F., Eds.; Academic Press: Cambridge, MA, USA, 2020; Volume 47, pp. 37–62.
- Schmidt, S.; Bornscheuer, U.T. Baeyer-Villiger monooxygenases: From protein engineering to biocatalytic applications. In Enzymes; Chaiyen, P., Tamanoi, F., Tamanoi, F., Eds.; Academic Press: Cambridge, MA, USA, 2020; Volume 47, pp. 231–281.
- de Gonzalo, G.; Mihovilovic, M.D.; Fraaije, M.W. Recent Developments in the Application of Baeyer-Villiger Monooxygenases as Biocatalysts. ChemBioChem 2010, 11, 2208–2231.
- Leisch, H.; Morley, K.; Lau, P.C.K. Baeyer-Villiger Monooxygenases: More Than Just Green Chemistry. Chem. Rev. 2011, 111, 4165–4222.
- Balke, K.; Kadow, M.; Mallin, H.; Sass, S.; Bornscheuer, U.T. Discovery, application and protein engineering of Baeyer-Villiger monooxygenases for organic synthesis. Org. Biomol. Chem. 2012, 10, 6249–6265.
- Bucko, M.; Gemeiner, P.; Schenkmayerova, A.; Krajcovic, T.; Rudroff, F.; Mihovilovic, M.D. Baeyer-Villiger oxidations: Biotechnological approach. Appl. Microbiol. Biotechnol. 2016, 100, 6585–6599.
- Knaus, T.; Toogood, H.S.; Scrutton, N.S. Ene-reductases and their Applications. In Green Biocatalysis; Patel, R.N., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 473–488.
- Furst, M.; Gran-Scheuch, A.; Aalbers, F.S.; Fraaije, M.W. Baeyer-Villiger Monooxygenases: Tunable Oxidative Biocatalysts. ACS Catal. 2019, 9, 11207–11241.
- Shi, Q.; Wang, H.; Liu, J.; Li, S.; Guo, J.; Li, H.; Jia, X.; Huo, H.; Zheng, Z.; You, S.; et al. Old yellow enzymes: Structures and structure-guided engineering for stereocomplementary bioreduction. Appl. Microbiol. Biotechnol. 2020, 104, 8155–8170.
- Huijbers, M.M.E.; Montersino, S.; Westphal, A.H.; Tischler, D.; van Berkel, W.J.H. Flavin dependent monooxygenases. Arch. Biochem. Biophys. 2014, 544, 2–17.
- Cummings Ryerson, C.; Ballou, D.P.; Walsh, C. Mechanistic Studies on Cyclohexanone Oxygenase. Biochemistry 1982, 21, 2644–2655.
- van Berkel, W.J.H.; Kamerbeek, N.M.; Fraaije, M.W. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 2006, 124, 670–689.
- Mthethwa, K.S.; Kassier, K.; Engel, J.; Kara, S.; Smit, M.S.; Opperman, D.J. Fungal BVMOs as alternatives to cyclohexanone monooxygenase. Enzym. Microb. Technol. 2017, 106, 11–17.
- van Beek, H.L.; Beyer, N.; Janssen, D.B.; Fraaije, M.W. Lyophilization conditions for the storage of monooxygenases. J. Biotechnol. 2015, 203, 41–44.
- Balke, K.; Beier, A.; Bornscheuer, U.T. Hot spots for the protein engineering of Baeyer-Villiger monooxygenases. Biotechnol. Adv. 2018, 36, 247–263.
- Fraaije, M.W.; Wu, J.; Heuts, D.P.H.M.; Van Hellemond, E.W.; Spelberg, J.H.L.; Janssen, D.B. Discovery of a thermostable Baeyer-Villiger monooxygenase by genome mining. Appl. Microbiol. Biotechnol. 2005, 66, 393–400.
- de Gonzalo, G.; Ottolina, G.; Zambianchi, F.; Fraaije, M.W.; Carrea, G. Biocatalytic properties of Baeyer-Villiger monooxygenases in aqueous-organic media. J. Mol. Catal. B Enzym. 2006, 39, 91–97.
- Rodríguez, C.; de Gonzalo, G.; Fraaije, M.W.; Gotor, V. Ionic liquids for enhancing the enantioselectivity of isolated BVMO-catalysed oxidations. Green Chem. 2010, 12, 2255–2260.
- Stewart, J.D. Cyclohexanone monooxygenase: A useful reagent for asymmetric baeyer-villiger reactions. Curr. Org. Chem. 1998, 2, 195–216.
- Secundo, F.; Fialà, S.; Fraaije, M.W.; De Gonzalo, G.; Meli, M.; Zambianchi, F.; Ottolina, G. Effects of water miscible organic solvents on the activity and conformation of the Baeyer-Villiger monooxygenases from Thermobifida fusca and Acinetobacter calcoaceticus: A comparative study. Biotechnol. Bioeng. 2011, 108, 491–499.
- Sartori, S.K.; Diaz, M.A.N.; Diaz-Muñoz, G. Lactones: Classification, synthesis, biological activities, and industrial applications. Tetrahedron 2021, 84, 132001.
- Wu, S.K.; Li, Z. Whole-Cell Cascade Biotransformations for One-Pot Multistep Organic Synthesis. ChemCatChem 2018, 10, 2164–2178.
- Fessner, W.D. Systems Biocatalysis: Development and engineering of cell-free “artificial metabolism” for preparative multi-enzymatic synthesis. New Biotech. 2015, 32, 658–664.
- Carballeira, J.D.; Quezada, M.A.; Hoyos, P.; Simeo, Y.; Hernaiz, M.J.; Alcantara, A.R.; Sinisterra, J.V. Microbial cells as catalysts for stereoselective red-ox reactions. Biotechnol. Adv. 2009, 27, 686–714.
- Kadisch, M.; Willrodt, C.; Hillen, M.; Buhler, B.; Schmid, A. Maximizing the stability of metabolic engineering-derived whole-cell biocatalysts. Biotechnol. J. 2017, 12, 29.
- Polakovic, M.; Svitel, J.; Bucko, M.; Filip, J.; Nedela, V.; Ansorge-Schumacher, M.B.; Gemeiner, P. Progress in biocatalysis with immobilized viable whole cells: Systems development, reaction engineering and applications. Biotechnol. Lett. 2017, 39, 667–683.
- Garzon-Posse, F.; Becerra-Figueroa, L.; Hernandez-Arias, J.; Gamba-Sanchez, D. Whole Cells as Biocatalysts in Organic Transformations. Molecules 2018, 23, 37.
- Musa, M.M.; Phillips, R.S. Recent advances in alcohol dehydrogenase-catalyzed asymmetric production of hydrophobic alcohols. Catal. Sci. Technolog. 2011, 1, 1311–1323.
- Kratzer, R.; Woodley, J.M.; Nidetzky, B. Rules for biocatalyst and reaction engineering to implement effective, NAD(P)H-dependent, whole cell bioreductions. Biotechnol. Adv. 2015, 33, 1641–1652.
- Zheng, Y.G.; Yin, H.H.; Yu, D.F.; Chen, X.; Tang, X.L.; Zhang, X.J.; Xue, Y.P.; Wang, Y.J.; Liu, Z.Q. Recent advances in biotechnological applications of alcohol dehydrogenases. Appl. Microbiol. Biotechnol. 2017, 101, 987–1001.
- Gonzalo, G.; Lavandera, I. Recent advances in selective biocatalytic (Hydrogen Transfer) reductions. In Homogeneous Hydrogenation with Non-Precious Catalysts; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2019; pp. 227–259.
- Willetts, A.J.; Knowles, C.J.; Levitt, M.S.; Roberts, S.M.; Sandey, H.; Shipston, N.F. Biotransformation of endo-bicyclo[2.2.1]heptan-2-ols and endo-bicyclo[3.2.0]-hept-2-en-6-ol into the corresponding lactones. J. Chem. Soc. Perkin Trans. 1 1991, 1608–1610.
- Lipik, V.T.; Kong, J.F.; Chattopadhyay, S.; Widjaja, L.K.; Liow, S.S.; Venkatraman, S.S.; Abadie, M.J.M. Thermoplastic biodegradable elastomers based on ε-caprolactone and L-lactide block co-polymers: A new synthetic approach. Acta Biomater. 2010, 6, 4261–4270.
- Nakayama, Y.; Aihara, K.; Yamanishi, H.; Fukuoka, H.; Tanaka, R.; Cai, Z.; Shiono, T. Synthesis of biodegradable thermoplastic elastomers from ε-caprolactone and lactide. J. Polym. Sci. Part A 2015, 53, 489–495.
- Schmidt, S.; Scherkus, C.; Muschiol, J.; Menyes, U.; Winkler, T.; Hummel, W.; Groger, H.; Liese, A.; Herz, H.G.; Bornscheuer, U.T. An Enzyme Cascade Synthesis of epsilon-Caprolactone and its Oligomers. Angew. Chem. Int. Ed. 2015, 54, 2784–2787.
- Schmidt, S.; Buchsenschutz, H.C.; Scherkus, C.; Liese, A.; Groger, H.; Bornscheuer, U.T. Biocatalytic Access to Chiral Polyesters by an Artificial Enzyme Cascade Synthesis. ChemCatChem 2015, 7, 3951–3955.
- Scherkus, C.; Schmidt, S.; Bornscheuer, U.T.; Groeger, H.; Kara, S.; Liese, A. Kinetic insights into e-caprolactone synthesis: Improvement of an enzymatic cascade reaction. Biotechnol. Bioeng. 2017, 114, 1215–1221.
- Kohl, A.; Srinivasamurthy, V.; Boettcher, D.; Kabisch, J.; Bornscheuer, U.T. Co-expression of an alcohol dehydrogenase and a cyclohexanone monooxygenase for cascade reactions facilitates the regeneration of the NADPH cofactor. Enz. Microb. Technol. 2018, 108, 53–58.
- Srinivasamurthy, V.S.T.; Bottcher, D.; Engel, J.; Kara, S.; Bornscheuer, U.T. A whole-cell process for the production of epsilon-caprolactone in aqueous media. Process Biochem. 2020, 88, 22–30.
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369.
- Jeon, E.Y.; Baek, A.H.; Bornscheuer, U.T.; Park, J.B. Enzyme fusion for whole-cell biotransformation of long-chain sec-alcohols into esters. Appl. Microbiol. Biotechnol. 2015, 99, 6267–6275.
- Toogood, H.S.; Scrutton, N.S. New developments in ‘ene’-reductase catalysed biological hydrogenations. Curr. Opin. Chem. Biol. 2014, 19, 107–115.
- Toogood, H.S.; Scrutton, N.S. Discovery, Characterization, Engineering, and Applications of Ene-Reductases for Industrial Biocatalysis. ACS Catal. 2018, 8, 3532–3549.
- Nett, N.; Duewel, S.; Schmermund, L.; Benary, G.E.; Ranaghan, K.; Mulholland, A.; Opperman, D.J.; Hoebenreich, S. A robust and stereocomplementary panel of ene-reductase variants for gram-scale asymmetric hydrogenation. Mol. Cat. 2021, 502, 111404.
- Silva, A.L.P.; Batista, P.K.; Filho, A.D.; do Nascimento Junior, C.S.; Reboucas, J.S.; Vale, J.A. Rapid conversion of cyclohexenone, cyclohexanone and cyclohexanol to epsilon-caprolactone by whole cells of Geotrichum candidum CCT 1205. Biocatal. Biotransfor. 2017, 35, 185–190.
- Mihovilovic, M.D.; Snajdrova, R.; Grotzl, B. Microbial Baeyer-Villiger oxidation of 4,4-disubstituted cyclohexan—And cyclohexenones by recombinant whole-cells expressing monooxygenases of bacterial origin. J. Mol. Catal. B Enzym. 2006, 39, 135–140.
- Silva, A.L.P.; Caridade, T.N.D.; Magalhaes, R.R.; de Sousa, K.T.; de Sousa, C.C.; Vale, J.A. Biocatalytic production of e-caprolactone using Geotrichum candidum cells immobilized on functionalized silica. Appl. Microbiol. Biotechnol. 2020, 104, 8887–8895.
- Liu, J.; Li, Z. Cascade Biotransformations via Enantioselective Reduction, Oxidation, and Hydrolysis: Preparation of (R)-delta-Lactones from 2-Alkylidenecyclopentanones. ACS Catal. 2013, 3, 908–911.
- Shin, J.; Martello, M.T.; Shrestha, M.; Wissinger, J.E.; Tolman, W.B.; Hillmyer, M.A. Pressure-sensitive adhesives from renewable triblock copolymers. Macromolecules 2011, 44, 87–94.
- Wanamaker, C.L.; O’Leary, L.E.; Lynd, N.A.; Hillmeyer, M.A.; Tolman, W.B. Renewable-resource thermoplastic elastomers based on polylactide and polymenthide. Biomacromolecules 2007, 8, 3634–3640.
- Gurusamy-Thangavelu, S.A.; Emond, S.J.; Kulshrestha, A.; Hillmyer, M.A.; MacOsko, C.W.; Tolman, W.B.; Hoye, T.R. Polyurethanes based on renewable polyols from bioderived lactones. Polym. Chem. 2012, 3, 2941–2948.
- Iqbal, N.; Stewart, J.D.; Macheroux, P.; Rudroff, F.; Mihovilovic, M.D. Novel concurrent redox cascades of (R)- and (S)-carvones enables access to carvo-lactones with distinct regio- and enantioselectivity. Tetrahedron 2018, 74, 7389–7394.
- Brigé, A.; Van Den Hemel, D.; Carpentier, W.; De Smet, L.; Van Beeumen, J.J. Comparative characterization and expression analysis of the four Old Yellow Enzyme homologues from Shewanella oneidensis indicate differences in physiological function. Biochem. J. 2006, 394, 335–344.
- Hall, M.; Stueckler, C.; Kroutil, W.; Macheroux, P.; Faber, K. Asymmetric bioreduction of activated alkenes using cloned 12-oxophytodienoate reductase isoenzymes OPR-1 and OPR-3 from Lycopersicon esculentum (tomato): A striking change of stereoselectivity. Angew. Chem. Int. Ed. 2007, 46, 3934–3937.
- Hall, M.; Stueckler, C.; Ehammer, H.; Pointner, E.; Oberdorfer, G.; Gruber, K.; Hauer, B.; Stuermer, R.; Kroutil, W.; Macheroux, P.; et al. Asymmetric bioreduction of C=C bonds using enoate reductases OPR1, OPR3 and YqjM: Enzyme-based stereocontrol. Adv. Synth. Catal. 2008, 350, 411–418.
- Padhi, S.K.; Bougioukou, D.J.; Stewart, J.D. Site-saturation mutagenesis of tryptophan 116 of Saccharomyces pastorianus old yellow enzyme uncovers stereocomplementary variants. J. Am. Chem. Soc. 2009, 131, 3271–3280.
- Donoghue, N.A.; Norris, D.B.; Trudgill, P.W. The Purification and Properties of Cyclohexanone Oxygenase from Nocardia globerula CL1 and Acinetobacter NCIB 9871. Eur. J. Biochem. 1976, 63, 175–192.
- Brzostowicz, P.C.; Gibson, K.L.; Thomas, S.M.; Blasko, M.S.; Rouvière, P.E. Simultaneous identification of two cyclohexanone oxidation genes from an environmental Brevibacterium isolate using MRNA differential display. J. Bacteriol. 2000, 182, 4241–4248.
- Griffin, M.; Trudgill, P.W. Purification and Properties of Cyclopentanone Oxygenase of Pseudomonas NCIB 9872. Eur. J. Biochem. 1976, 63, 199–209.
- Krow, G.R. The Baeyer–Villiger Oxidation of Ketones and Aldehydes. In Organic Reactions; Paquette, L.A., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1993; Volume 43, pp. 251–353.
- Oberleitner, N.; Ressmann, A.K.; Bica, K.; Gartner, P.; Fraaije, M.W.; Bornscheuer, U.T.; Rudroff, F.; Mihovilovic, M.D. From waste to value—Direct utilization of limonene from orange peel in a biocatalytic cascade reaction towards chiral carvolactone. Green Chem. 2017, 19, 367–371.
- Oberleitner, N.; Peters, C.; Muschiol, J.; Kadow, M.; Sass, S.; Bayer, T.; Schaaf, P.; Iqbal, N.; Rudroff, F.; Mihovilovic, M.D.; et al. An Enzymatic Toolbox for Cascade Reactions: A Showcase for an In Vivo Redox Sequence in Asymmetric Synthesis. ChemCatChem 2013, 5, 3524–3528.
- Avalos, G.A.A.; Toogood, H.S.; Tait, S.; Messiha, H.L.; Scrutton, N.S. From Bugs to Bioplastics: Total (+)-Dihydrocarvide Biosynthesis by Engineered Escherichia coli. ChemBioChem 2019, 20, 785–792.
- Fryszkowska, A.; Toogood, H.; Sakuma, M.; Gardiner, J.M.; Stephens, G.M.; Scrutton, N.S. Asymmetrie reduction of activated alkenes by pentaerythritol tetranitrate reductase: Specificity and control of stereochemical outcome by reaction optimisation. Adv. Synth. Catal. 2009, 351, 2976–2990.
- French, C.E.; Nicklin, S.; Bruce, N.C. Sequence and properties of pentaerythritol tetranitrate reductase from Enterobacter cloacae PB2. J. Bacteriol. 1996, 178, 6623–6627.
- Messiha, H.L.; Ahmed, S.T.; Karuppiah, V.; Suardíaz, R.; Ascue Avalos, G.A.; Fey, N.; Yeates, S.; Toogood, H.S.; Mulholland, A.J.; Scrutton, N.S. Biocatalytic Routes to Lactone Monomers for Polymer Production. Biochemistry 2018, 57, 1997–2008.