Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Genetics & Heredity
Mutations in DNA can be limited to one or a few nucleotides, or encompass larger deletions, insertions, duplications, inversions and translocations that span long stretches of DNA or even full chromosomes. These so-called structural variations (SVs) can alter the gene copy number, modify open reading frames, change regulatory sequences or chromatin structure and thus result in major phenotypic changes.
- structural variation
- fungi
- adaptation
1. Introduction
Structural variation (SV) groups different forms of mutations that involve longer stretches of DNA, including deletions, insertions, duplications, inversions, translocations, or even full chromosome fusion, fission or loss (Figure 1). Structural variants can be balanced and show no specific loss or gain of DNA information, such as inversions of a genetic fragment or translocations of a stretch of DNA within or between chromosomes, or they can be unbalanced, where a part of the genome is lost (deletions), acquired (insertions) or duplicated (duplications), which is termed copy number variation (CNV).
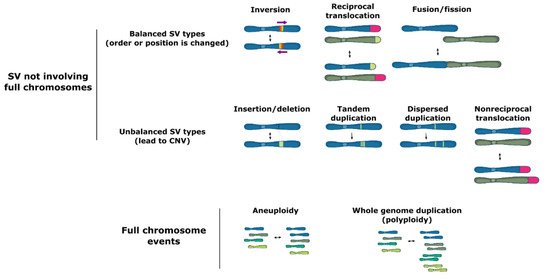
Figure 1. Types of structural variation.
Structural variation may occur both in coding and noncoding regions of the genome, including in highly repetitive elements, such as transposons. SV events can lead to major phenotypic changes via diverse mechanisms including modification of open reading frames, changes in gene expression due to copy number variation, alteration of regulatory sequences (via gain or loss of functional genomic elements) or chromatin structure, or even formation of novel genes [1][2][3][4][5]. Moreover, some forms of SV, such as large inversions and chromosomal fusions, cause a reduction in recombination rates between homologous chromosome pairs. In turn, the reduced recombination may facilitate the cosegregation of multiple adaptive polymorphisms as if they were controlled by a single genetic locus (linkage disequilibrium and supergene formation) [6][7][8][9][10][11].
In humans, single nucleotide variants (SNVs) are the most common type of variation, but SV accounts for a higher number of variable nucleotides between genomes, with roughly 0.5% of the human genome being involved in structural variation [12][13]. Strikingly, third-generation (long-read) genome sequencing of a clonal population of seven closely related Schizosaccharomyces pombe strains that diverged ∼50–65 years ago revealed that they have an average pairwise difference of 19 SNVs and four nonoverlapping larger duplications [14]. Moreover, SVs are three times more likely to be associated with a genome-wide association signal and 50 times more likely to be associated with expressed quantitative trait loci than single nucleotide variants, further hinting at their importance as drivers of phenotypic variation [13][15]. Importantly, despite the significant contribution of SV events (especially of CNVs) to quantitative traits, they are frequently overlooked in studies employing short-read sequencing technologies [14].
The phenotypic consequences of SVs have traditionally been assumed to be almost exclusively negative. This is perhaps partly due to the association of SVs with many human diseases, especially autoimmune, metabolic, and cognitive disorders [16][17][18][19][20]. However, the emergence of advanced genotype-to-phenotype mapping technologies, as well as studies focusing on experimental evolution have led to a growing body of evidence suggesting that many SVs are neutral or even adaptive, both in humans [12][15][21] and other organisms, including microbes [11][22][23][24][25][26][27][28][29][30][31]. SVs are therefore increasingly considered to be an important evolutionary driver, and some studies suggest that SV may be especially important for quick adaptation.
2. Mechanisms of SV Formation
SV involving complete chromosomes is often caused by defective chromosome segregation. Chromosomes must be meticulously replicated and equally segregated at each cell division. Distortion of either one of these processes can lead to SV formation. In particular, failure of any of the critical chromosome segregation steps, including chromatid cohesion, spindle pole body (functional equivalent of the mammalian centrosome) formation at opposite cell poles, kinetochore–microtubule attachment, and quality control at the spindle assembly checkpoint can result in aneuploidy (i.e., loss or gain of whole chromosomes) (Figure 2A) [32].
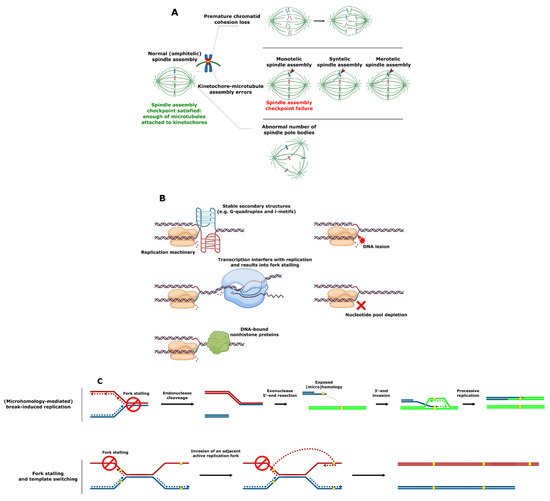
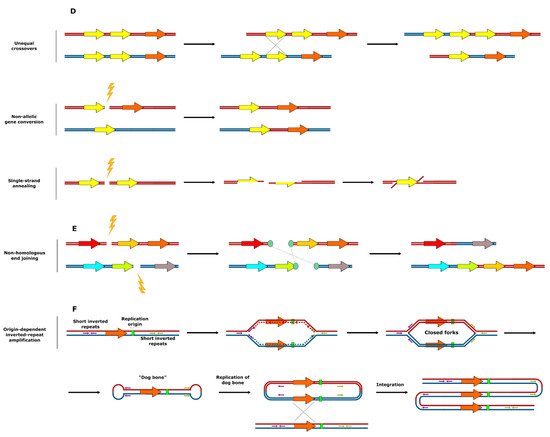
Figure 2. Mechanisms of SV formation. (A) Events leading to aneuploidy. (B) Events leading to replication fork collapse. (C) SV formation as a result of stalled replication fork reactivation. (D) SV formation mediated by homologous recombination. (E) SV formation mediated by nonhomologous end joining. (F) Origin-dependent inverted-repeat amplification.
An SV that does not involve full chromosomes often results from compromised DNA replication, where processive forks collide with the replication fork barriers (Figure 2B) [33][34][35]. These barriers typically include (1) specific DNA secondary structures such as G-quadruplex (G4) motifs [36][37][38], which are enriched in the telomeres, ribosomal DNA (rDNA) and promoter regions in S. cerevisiae, Schizosaccharomyces pombe, and human cells [39][40][41][42][43][44]; (2) highly expressed loci such as the tRNA genes where transcription can interfere with replication [45][46][47]; or (3) tightly DNA bound nonhistone proteins (e.g., at centromeres) [48][49]. Replication forks can also be stalled as a result of DNA damage or the inhibition of replication by nucleotide depletion [50][51]. Reactivation of blocked replication forks and DNA damage can lead to SV due to the occurrence of nonallelic homologous recombination resulting from incorrect repair template utilization (Figure 2C) [52][53][54]. This process is remarkably more frequent in the case of dispersed repetitive DNA sequences such as transposable elements or remnants of those (long terminal repeats), tRNA genes, origins of replication, and clusters of tandemly repeated genes including those encoding ribosomal RNA and those residing in subtelomeric duplication blocks [14][54][55][56][57][58][59][60][61][62][63][64][65][66][67]. Curiously, stretches of repetitive DNA and, in particular, transposable elements are enriched in highly fast-evolving genomic compartments (which exist as ‘islands’ on core chromosomes) and accessory chromosomes of many pathogenic filamentous fungi [68][69][70][71][72][73][74]. These genomic compartments were shown to be the hot spots of SV [73][74]. Increased plasticity of the indicated genomic regions known to bear the virulence-related genes likely allows pathogens to keep up with the evolution of the host defense mechanisms and succeed in pathogen–host “arms race”.
A third major mechanism underlying SV is linked to crossing-over between repetitive DNA sequences and the repair of DNA double strand breaks near repetitive DNA sequences (Figure 2D,E) [75][76]. Various types of homologous recombination at repeat sites, including unequal crossovers, gene conversion, and single-strand annealing were reported to result in CNV [75]. A specific example of repeat-associated CNV generation, origin-dependent inverted-repeat amplification (Figure 2F), was hypothesized to underlie the amplification of the SUL1 locus in yeast [77][78]. As a result of the DNA replication error at small, interrupted inverted repeats, nascent leading and lagging strands get covalently linked. This ends up in formation of an extrachromosomal circular intermediate, and its integration into original chromosomal locus results in the gene triplication [77][78]. In some specific cases, copy number amplification is achieved via the formation of the extrachromosomal circular elements [79], which were proposed to be a fast and revocable mechanism of gene copy number amplification [80][81].
Finally, a very specific source of gene duplications is the whole genome duplication (WGD, also referred to as polyploidization)—i.e., addition of a complete set of chromosomes to the genome [82][83].
3. Balanced SV Events
Balanced SV types such as reciprocal translocations and inversions are widespread in Saccharomyces species and other fungi [14][60][63][65][71][72][84][85][86][87][88][89]. They are thought to serve as initial genetic barriers in eukaryotic speciation and, thus, to contribute to the onset of reproductive isolation and speciation [90][91][92][93][94][95][96]. In flies [97], mosquitoes [98], and flowering plants [8], inversions are hypothesized to also play a role in evolutionary adaptation. Analysis of the outcomes of chromosomal translocations in S. cerevisiae [99] and of translocations and inversions in S. pombe [85][100] demonstrated that these types of SV can significantly influence the fitness of the organism in specific environments, possibly as some events cause changes in gene expression [85]. It was hypothesized that balanced types of SV can be maintained as polymorphisms in nature despite their meiotic costs (low viability in heterozygotic crosses) when this disadvantage is outweighed by the fitness advantage gained in mitosis (antagonistic pleiotropy) [85]. Contrastingly, Naseeb and colleagues were not able to detect phenotypic consequences of a set of large inversions, even if they did observe significant changes in gene transcription patterns [101]. This again underscores that the effect of a specific structural rearrangement always depends on the affected genetic locus, the genetic background and the environment.
This entry is adapted from the peer-reviewed paper 10.3390/genes12050699
References
- Stewart, N.B.; Rogers, R.L. Chromosomal rearrangements as a source of new gene formation in Drosophila yakuba. PLoS Genet. 2019, 15, e1008314.
- Huang, Y.C.; Dang, V.D.; Chang, N.C.; Wang, J. Multiple large inversions and breakpoint rewiring of gene expression in the evolution of the fire ant social supergene. Proc. Biol. Sci. 2018, 285.
- Lavington, E.; Kern, A.D. The Effect of Common Inversion Polymorphisms on Patterns of Transcriptional Variation in Drosophila melanogaster. G3 Genes Genomes Genet. 2017, 7, 3659.
- Gamazon, E.R.; Stranger, B.E. The impact of human copy number variation on gene expression. Brief. Funct. Genom. 2015, 14, 352–357.
- Radke, D.W.; Lee, C. Adaptive potential of genomic structural variation in human and mammalian evolution. Brief. Funct. Genom. 2015, 14, 358–368.
- Thompson, M.J.; Jiggins, C.D. Supergenes and their role in evolution. Heredity 2014, 113, 1–8.
- Avril, A.; Purcell, J.; Brelsford, A.; Chapuisat, M. Asymmetric assortative mating and queen polyandry are linked to a supergene controlling ant social organization. Mol. Ecol. 2019, 28, 1428–1438.
- Coughlan, J.M.; Willis, J.H. Dissecting the role of a large chromosomal inversion in life history divergence throughout the Mimulus guttatus species complex. Mol. Ecol. 2019, 28, 1343–1357.
- Faria, R.; Chaube, P.; Morales, H.E.; Larsson, T.; Lemmon, A.R.; Lemmon, E.M.; Rafajlović, M.; Panova, M.; Ravinet, M.; Johannesson, K.; et al. Multiple chromosomal rearrangements in a hybrid zone between Littorina saxatilis ecotypes. Mol. Ecol. 2019, 28, 1375–1393.
- Wellband, K.; Mérot, C.; Linnansaari, T.; Elliott, J.A.K.; Curry, R.A.; Bernatchez, L. Chromosomal fusion and life history-associated genomic variation contribute to within-river local adaptation of Atlantic salmon. Mol. Ecol. 2019, 28, 1439–1459.
- Wellenreuther, M.; Mérot, C.; Berdan, E.; Bernatchez, L. Going beyond SNPs: The role of structural genomic variants in adaptive evolution and species diversification. Mol. Ecol. 2019, 28, 1203–1209.
- Conrad, D.F.; Pinto, D.; Redon, R.; Feuk, L.; Gokcumen, O.; Zhang, Y.; Aerts, J.; Andrews, T.D.; Barnes, C.; Campbell, P.; et al. Origins and functional impact of copy number variation in the human genome. Nature 2010, 464, 704–712.
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.; et al. An integrated map of structural variation in 2504 human genomes. Nature 2015, 526, 75–81.
- Jeffares, D.C.; Jolly, C.; Hoti, M.; Speed, D.; Shaw, L.; Rallis, C.; Balloux, F.; Dessimoz, C.; Bähler, J.; Sedlazeck, F.J. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 2017, 8, 14061.
- Audano, P.A.; Sulovari, A.; Graves-Lindsay, T.A.; Cantsilieris, S.; Sorensen, M.; Welch, A.E.; Dougherty, M.L.; Nelson, B.J.; Shah, A.; Dutcher, S.K.; et al. Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 2019, 176, 663–675.e619.
- Weischenfeldt, J.; Symmons, O.; Spitz, F.; Korbel, J.O. Phenotypic impact of genomic structural variation: Insights from and for human disease. Nat. Rev. Genet. 2013, 14, 125–138.
- Hollox, E.J.; Huffmeier, U.; Zeeuwen, P.L.J.M.; Palla, R.; Lascorz, J.; Rodijk-Olthuis, D.; van de Kerkhof, P.C.M.; Traupe, H.; de Jongh, G.; Heijer, M.D.; et al. Psoriasis is associated with increased β-defensin genomic copy number. Nat. Genet. 2008, 40, 23–25.
- Stefansson, H.; Rujescu, D.; Cichon, S.; Pietilainen, O.P.; Ingason, A.; Steinberg, S.; Fossdal, R.; Sigurdsson, E.; Sigmundsson, T.; Buizer-Voskamp, J.E.; et al. Large recurrent microdeletions associated with schizophrenia. Nature 2008, 455, 232–236.
- Traherne, J.A.; Martin, M.; Ward, R.; Ohashi, M.; Pellett, F.; Gladman, D.; Middleton, D.; Carrington, M.; Trowsdale, J. Mechanisms of copy number variation and hybrid gene formation in the KIR immune gene complex. Hum. Mol. Genet. 2010, 19, 737–751.
- Polley, S.; Louzada, S.; Forni, D.; Sironi, M.; Balaskas, T.; Hains, D.S.; Yang, F.; Hollox, E.J. Evolution of the rapidly mutating human salivary agglutinin gene (DMBT1) and population subsistence strategy. Proc. Natl. Acad. Sci. USA 2015, 112, 5105–5110.
- Durbin, R.M.; Altshuler, D.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Collins, F.S.; De La Vega, F.M.; Donnelly, P.; et al. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073.
- Weissensteiner, M.H.; Bunikis, I.; Catalan, A.; Francoijs, K.J.; Knief, U.; Heim, W.; Peona, V.; Pophaly, S.D.; Sedlazeck, F.J.; Suh, A.; et al. Discovery and population genomics of structural variation in a songbird genus. Nat. Commun. 2020, 11, 3403.
- Catanach, A.; Crowhurst, R.; Deng, C.; David, C.; Bernatchez, L.; Wellenreuther, M. The genomic pool of standing structural variation outnumbers single nucleotide polymorphism by threefold in the marine teleost Chrysophrys auratus. Mol. Ecol. 2019, 28, 1210–1223.
- Lucek, K.; Gompert, Z.; Nosil, P. The role of structural genomic variants in population differentiation and ecotype formation in Timema cristinae walking sticks. Mol. Ecol. 2019, 28, 1224–1237.
- Chakraborty, M.; Emerson, J.J.; Macdonald, S.J.; Long, A.D. Structural variants exhibit widespread allelic heterogeneity and shape variation in complex traits. Nat. Commun. 2019, 10, 4872.
- Fisher, K.J.; Buskirk, S.W.; Vignogna, R.C.; Marad, D.A.; Lang, G.I. Adaptive genome duplication affects patterns of molecular evolution in Saccharomyces cerevisiae. PLoS Genet. 2018, 14, e1007396.
- Steenwyk, J.; Rokas, A. Extensive Copy Number Variation in Fermentation-Related Genes among Saccharomyces cerevisiae Wine Strains. G3 Genes Genomes Genet. 2017, 7, 1475.
- Zhang, K.; Zhang, L.J.; Fang, Y.H.; Jin, X.N.; Qi, L.; Wu, X.C.; Zheng, D.Q. Genomic structural variation contributes to phenotypic change of industrial bioethanol yeast Saccharomyces cerevisiae. FEMS Yeast Res. 2016, 16, 118.
- Treu, L.; Toniolo, C.; Nadai, C.; Sardu, A.; Giacomini, A.; Corich, V.; Campanaro, S. The impact of genomic variability on gene expression in environmental Saccharomyces cerevisiae strains. Environ. Microbiol. 2014, 16, 1378–1397.
- Gresham, D.; Desai, M.M.; Tucker, C.M.; Jenq, H.T.; Pai, D.A.; Ward, A.; DeSevo, C.G.; Botstein, D.; Dunham, M.J. The Repertoire and Dynamics of Evolutionary Adaptations to Controlled Nutrient-Limited Environments in Yeast. PLoS Genet. 2008, 4, e1000303.
- Dunham, M.J.; Badrane, H.; Ferea, T.; Adams, J.; Brown, P.O.; Rosenzweig, F.; Botstein, D. Characteristic genome rearrangements in experimental evolution of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2002, 99, 16144.
- Thompson, S.L.; Bakhoum, S.F.; Compton, D.A. Mechanisms of chromosomal instability. Curr. Biol. 2010, 20, R285–R295.
- Myung, K.; Datta, A.; Kolodner, R.D. Suppression of spontaneous chromosomal rearrangements by S phase checkpoint functions in Saccharomyces cerevisiae. Cell 2001, 104, 397–408.
- Labib, K.; Hodgson, B. Replication fork barriers: Pausing for a break or stalling for time? EMBO Rep. 2007, 8, 346–353.
- Admire, A.; Shanks, L.; Danzl, N.; Wang, M.; Weier, U.; Stevens, W.; Hunt, E.; Weinert, T. Cycles of chromosome instability are associated with a fragile site and are increased by defects in DNA replication and checkpoint controls in yeast. Genes Dev. 2006, 20, 159–173.
- Brewer, B.J.; Lockshon, D.; Fangman, W.L. The arrest of replication forks in the rDNA of yeast occurs independently of transcription. Cell 1992, 71, 267–276.
- Paeschke, K.; Bochman, M.L.; Garcia, P.D.; Cejka, P.; Friedman, K.L.; Kowalczykowski, S.C.; Zakian, V.A. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature 2013, 497, 458–462.
- Wallgren, M.; Mohammad, J.B.; Yan, K.-P.; Pourbozorgi-Langroudi, P.; Ebrahimi, M.; Sabouri, N. G-rich telomeric and ribosomal DNA sequences from the fission yeast genome form stable G-quadruplex DNA structures in vitro and are unwound by the Pfh1 DNA helicase. Nucleic Acids Res. 2016, 44, 6213–6231.
- Capra, J.A.; Paeschke, K.; Singh, M.; Zakian, V.A. G-quadruplex DNA sequences are evolutionarily conserved and associated with distinct genomic features in Saccharomyces cerevisiae. PLoS Comput. Biol. 2010, 6, e1000861.
- Hershman, S.G.; Chen, Q.; Lee, J.Y.; Kozak, M.L.; Yue, P.; Wang, L.S.; Johnson, F.B. Genomic distribution and functional analyses of potential G-quadruplex-forming sequences in Saccharomyces cerevisiae. Nucleic Acids Res. 2008, 36, 144–156.
- Huppert, J.L.; Balasubramanian, S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005, 33, 2908–2916.
- Sabouri, N.; Capra, J.A.; Zakian, V.A. The essential Schizosaccharomyces pombe Pfh1 DNA helicase promotes fork movement past G-quadruplex motifs to prevent DNA damage. BMC Biol. 2014, 12, 1–14.
- Huppert, J.L.; Balasubramanian, S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007, 35, 406–413.
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P. Anticancer activity of CX-3543: A direct inhibitor of rRNA biogenesis. Cancer Res. 2009, 69, 7653–7661.
- Ivessa, A.S.; Lenzmeier, B.A.; Bessler, J.B.; Goudsouzian, L.K.; Schnakenberg, S.L.; Zakian, V.A. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol. Cell 2003, 12, 1525–1536.
- Lin, Y.-L.; Pasero, P. Interference between DNA replication and transcription as a cause of genomic instability. Curr. Genom. 2012, 13, 65–73.
- Gaillard, H.; Aguilera, A. Transcription as a threat to genome integrity. Annu. Rev. Biochem. 2016, 85, 291–317.
- Cabral, M.; Cheng, X.; Singh, S.; Ivessa, A.S. Absence of Non-histone Protein Complexes at Natural Chromosomal Pause Sites Results in Reduced Replication Pausing in Aging Yeast Cells. Cell Rep. 2016, 17, 1747–1754.
- Lambert, S.; Watson, A.; Sheedy, D.M.; Martin, B.; Carr, A.M. Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell 2005, 121, 689–702.
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446.
- Saldivar, J.C.; Miuma, S.; Bene, J.; Hosseini, S.A.; Shibata, H.; Sun, J.; Wheeler, L.J.; Mathews, C.K.; Huebner, K. Initiation of genome instability and preneoplastic processes through loss of Fhit expression. PLoS Genet. 2012, 8, e1003077.
- Lambert, S.; Mizuno, K.I.; Blaisonneau, J.; Martineau, S.; Chanet, R.; Fréon, K.; Murray, J.M.; Carr, A.M.; Baldacci, G. Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol. Cell 2010, 39, 346–359.
- Mizuno, K.I.; Lambert, S.; Baldacci, G.; Murray, J.M.; Carr, A.M. Nearby inverted repeats fuse to generate acentric and dicentric palindromic chromosomes by a replication template exchange mechanism. Genes Dev. 2009, 23, 2876–2886.
- Payen, C.; Koszul, R.; Dujon, B.; Fischer, G. Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet. 2008, 4, e1000175.
- Petes, T.D. Unequal meiotic recombination within tandem arrays of yeast ribosomal DNA genes. Cell 1980, 19, 765–774.
- Szostak, J.W.; Wu, R. Unequal crossing over in the ribosomal DNA of Saccharomyces cerevisiae. Nature 1980, 284, 426–430.
- Welch, J.W.; Maloney, D.H.; Fogel, S. Unequal crossing-over and gene conversion at the amplified CUP1 locus of yeast. Mol. Gen. Genet. MGG 1990, 222, 304–310.
- Gangloff, S.; Zou, H.; Rothstein, R. Gene conversion plays the major role in controlling the stability of large tandem repeats in yeast. EMBO J. 1996, 15, 1715–1725.
- Ozenberger, B.A.; Roeder, G.S. A unique pathway of double-strand break repair operates in tandemly repeated genes. Mol. Cell. Biol. 1991, 11, 1222–1231.
- Fischer, G.; James, S.; Roberts, I.; Oliver, S.; Louis, E. Chromosomal evolution in Saccharomyces. Nature 2000, 405, 451–454.
- Stankiewicz, P.; Lupski, J.R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002, 18, 74–82.
- Kellis, M.; Patterson, N.; Endrizzi, M.; Birren, B.; Lander, E.S. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 2003, 423, 241–254.
- Gordon, J.L.; Byrne, K.P.; Wolfe, K.H. Additions, losses, and rearrangements on the evolutionary route from a reconstructed ancestor to the modern Saccharomyces cerevisiae genome. PLoS Genet. 2009, 5, e1000485.
- Selmecki, A.M.; Maruvka, Y.E.; Richmond, P.A.; Guillet, M.; Shoresh, N.; Sorenson, A.L.; De, S.; Kishony, R.; Michor, F.; Dowell, R. Polyploidy can drive rapid adaptation in yeast. Nature 2015, 519, 349–352.
- Yue, J.-X.; Li, J.; Aigrain, L.; Hallin, J.; Persson, K.; Oliver, K.; Bergström, A.; Coupland, P.; Warringer, J.; Lagomarsino, M.C.; et al. Contrasting evolutionary genome dynamics between domesticated and wild yeasts. Nat. Genet. 2017, 49, 913–924.
- Sui, Y.; Qi, L.; Wu, J.-K.; Wen, X.-P.; Tang, X.-X.; Ma, Z.-J.; Wu, X.-C.; Zhang, K.; Kokoska, R.J.; Zheng, D.-Q.; et al. Genome-wide mapping of spontaneous genetic alterations in diploid yeast cells. Proc. Natl. Acad. Sci. USA 2020, 117, 28191.
- Kaya, A.; Mariotti, M.; Tyshkovskiy, A.; Zhou, X.; Hulke, M.L.; Ma, S.; Gerashchenko, M.V.; Koren, A.; Gladyshev, V.N. Molecular signatures of aneuploidy-driven adaptive evolution. Nat. Commun. 2020, 11, 588.
- Raffaele, S.; Farrer, R.A.; Cano, L.M.; Studholme, D.J.; MacLean, D.; Thines, M.; Jiang, R.H.Y.; Zody, M.C.; Kunjeti, S.G.; Donofrio, N.M.; et al. Genome Evolution Following Host Jumps in the Irish Potato Famine Pathogen Lineage. Science 2010, 330, 1540.
- Ma, L.-J.; van der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.-J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373.
- Klosterman, S.J.; Subbarao, K.V.; Kang, S.; Veronese, P.; Gold, S.E.; Thomma, B.P.; Chen, Z.; Henrissat, B.; Lee, Y.-H.; Park, J. Comparative genomics yields insights into niche adaptation of plant vascular wilt pathogens. PLoS Pathog. 2011, 7, e1002137.
- Chuma, I.; Isobe, C.; Hotta, Y.; Ibaragi, K.; Futamata, N.; Kusaba, M.; Yoshida, K.; Terauchi, R.; Fujita, Y.; Nakayashiki, H. Multiple translocation of the AVR-Pita effector gene among chromosomes of the rice blast fungus Magnaporthe oryzae and related species. PLoS Pathog. 2011, 7, e1002147.
- De Jonge, R.; Bolton, M.D.; Kombrink, A.; van den Berg, G.C.M.; Yadeta, K.A.; Thomma, B.P.H.J. Extensive chromosomal reshuffling drives evolution of virulence in an asexual pathogen. Genome Res. 2013, 23, 1271–1282.
- Faino, L.; Seidl, M.F.; Shi-Kunne, X.; Pauper, M.; van den Berg, G.C.; Wittenberg, A.H.; Thomma, B.P. Transposons passively and actively contribute to evolution of the two-speed genome of a fungal pathogen. Genome Res. 2016, 26, 1091–1100.
- Plissonneau, C.; Stürchler, A.; Croll, D. The Evolution of Orphan Regions in Genomes of a Fungal Pathogen of Wheat. mBio 2016, 7, e01231-16.
- Hastings, P.J.; Lupski, J.R.; Rosenberg, S.M.; Ira, G. Mechanisms of change in gene copy number. Nat. Rev. Genet. 2009, 10, 551–564.
- Carvalho, C.M.; Lupski, J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016, 17, 224–238.
- Brewer, B.J.; Payen, C.; Raghuraman, M.K.; Dunham, M.J. Origin-dependent inverted-repeat amplification: A replication-based model for generating palindromic amplicons. PLoS Genet. 2011, 7, e1002016.
- Brewer, B.J.; Payen, C.; Di Rienzi, S.C.; Higgins, M.M.; Ong, G.; Dunham, M.J.; Raghuraman, M.K. Origin-Dependent Inverted-Repeat Amplification: Tests of a Model for Inverted DNA Amplification. PLoS Genet. 2015, 11, e1005699.
- Gresham, D.; Usaite, R.; Germann, S.M.; Lisby, M.; Botstein, D.; Regenberg, B. Adaptation to diverse nitrogen-limited environments by deletion or extrachromosomal element formation of the GAP1 locus. Proc. Natl. Acad. Sci. USA 2010, 107, 18551–18556.
- Møller, H.D.; Andersen, K.S.; Regenberg, B. A model for generating several adaptive phenotypes from a single genetic event: Saccharomyces cerevisiae GAP1 as a potential bet-hedging switch. Commun. Integr. Biol. 2013, 6, e23933.
- Cohen, S.; Segal, D. Extrachromosomal circular DNA in eukaryotes: Possible involvement in the plasticity of tandem repeats. Cytogenet. Genome Res. 2009, 124, 327–338.
- Pontes, O.; Neves, N.; Silva, M.; Lewis, M.S.; Madlung, A.; Comai, L.; Viegas, W.; Pikaard, C.S. Chromosomal locus rearrangements are a rapid response to formation of the allotetraploid Arabidopsis suecica genome. Proc. Natl. Acad. Sci. USA 2004, 101, 18240–18245.
- Madlung, A.; Tyagi, A.P.; Watson, B.; Jiang, H.; Kagochi, T.; Doerge, R.W.; Martienssen, R.; Comai, L. Genomic changes in synthetic Arabidopsis polyploids. Plant J. 2005, 41, 221–230.
- Bendixsen, D.P.; Gettle, N.; Gilchrist, C.; Zhang, Z.; Stelkens, R. Genomic evidence of an ancient East Asian divergence event in wild Saccharomyces cerevisiae. Genome Biol. Evol. 2021, 13.
- Avelar, A.T.; Perfeito, L.; Gordo, I.; Ferreira, M.G. Genome architecture is a selectable trait that can be maintained by antagonistic pleiotropy. Nat. Commun. 2013, 4, 1–10.
- Gordon, J.L.; Byrne, K.P.; Wolfe, K.H. Mechanisms of chromosome number evolution in yeast. PLoS Genet. 2011, 7, e1002190.
- Fraser, J.A.; Huang, J.C.; Pukkila-Worley, R.; Alspaugh, J.A.; Mitchell, T.G.; Heitman, J. Chromosomal Translocation and Segmental Duplication in Cryptococcus neoformans. Eukaryotic Cell 2005, 4, 401.
- Bradshaw, R.E.; Sim, A.D.; Chettri, P.; Dupont, P.Y.; Guo, Y.; Hunziker, L.; McDougal, R.L.; van der Nest, A.; Fourie, A.; Wheeler, D. Global population genomics of the forest pathogen Dothistroma septosporum reveal chromosome duplications in high dothistromin-producing strains. Mol. Plant Pathol. 2019, 20, 784–799.
- Wang, Q.; Sun, M.; Zhang, Y.; Song, Z.; Zhang, S.; Zhang, Q.; Xu, J.R.; Liu, H. Extensive chromosomal rearrangements and rapid evolution of novel effector superfamilies contribute to host adaptation and speciation in the basal ascomycetous fungi. Mol. Plant Pathol. 2020, 21, 330–348.
- Hou, J.; Friedrich, A.; de Montigny, J.; Schacherer, J. Chromosomal rearrangements as a major mechanism in the onset of reproductive isolation in Saccharomyces cerevisiae. Curr. Biol. 2014, 24, 1153–1159.
- Noor, M.A.; Grams, K.L.; Bertucci, L.A.; Reiland, J. Chromosomal inversions and the reproductive isolation of species. Proc. Natl. Acad. Sci. USA 2001, 98, 12084–12088.
- Rieseberg, L.H. Chromosomal rearrangements and speciation. Trends Ecol. Evol. 2001, 16, 351–358.
- Kirkpatrick, M.; Barton, N. Chromosome inversions, local adaptation and speciation. Genetics 2006, 173, 419–434.
- Navarro, A.; Barton, N.H. Accumulating postzygotic isolation genes in parapatry: A new twist on chromosomal speciation. Evolution 2003, 57, 447–459.
- Seoighe, C.; Federspiel, N.; Jones, T.; Hansen, N.; Bivolarovic, V.; Surzycki, R.; Tamse, R.; Komp, C.; Huizar, L.; Davis, R.W.; et al. Prevalence of small inversions in yeast gene order evolution. Proc. Natl. Acad. Sci. USA 2000, 97, 14433–14437.
- Faria, R.; Navarro, A. Chromosomal speciation revisited: Rearranging theory with pieces of evidence. Trends Ecol. Evol. 2010, 25, 660–669.
- Puig Giribets, M.; García Guerreiro, M.P.; Santos, M.; Ayala, F.J.; Tarrío, R.; Rodríguez-Trelles, F. Chromosomal inversions promote genomic islands of concerted evolution of Hsp70 genes in the Drosophila subobscura species subgroup. Mol. Ecol. 2019, 28, 1316–1332.
- Ayala, D.; Zhang, S.; Chateau, M.; Fouet, C.; Morlais, I.; Costantini, C.; Hahn, M.W.; Besansky, N.J. Association mapping desiccation resistance within chromosomal inversions in the African malaria vector Anopheles gambiae. Mol. Ecol. 2019, 28, 1333–1342.
- Colson, I.; Delneri, D.; Oliver, S.G. Effects of reciprocal chromosomal translocations on the fitness of Saccharomyces cerevisiae. EMBO Rep. 2004, 5, 392–398.
- Brown, W.R.; Liti, G.; Rosa, C.; James, S.; Roberts, I.; Robert, V.; Jolly, N.; Tang, W.; Baumann, P.; Green, C.; et al. A Geographically Diverse Collection of Schizosaccharomyces pombe Isolates Shows Limited Phenotypic Variation but Extensive Karyotypic Diversity. G3 2011, 1, 615–626.
- Naseeb, S.; Carter, Z.; Minnis, D.; Donaldson, I.; Zeef, L.; Delneri, D. Widespread Impact of Chromosomal Inversions on Gene Expression Uncovers Robustness via Phenotypic Buffering. Mol. Biol. Evol. 2016, 33, 1679–1696.
This entry is offline, you can click here to edit this entry!