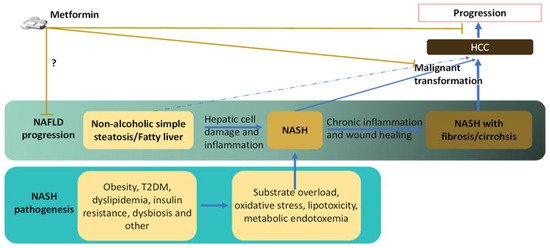
Nonalcoholic fatty liver disease (NAFLD) is strongly linked to the global epidemic of obesity and type 2 diabetes mellitus (T2DM). Notably, NAFLD can progress from the mildest form of simple steatosis to nonalcoholic steatohepatitis (NASH) that increases the risk for hepatocellular carcinoma (HCC), which is a malignancy with a dismal prognosis and rising incidence in the United States and other developed counties, possibly due to the epidemic of NAFLD. Metformin, the first-line drug for T2DM, has been suggested to reduce risks for several types of cancers including HCC and protect against NASH-related HCC, as revealed by epidemical studies on humans and preclinical studies on animal models.
- metformin
- NAFLD
- NASH
- HCC
- type 2 diabetes mellitus
- macrophage
- T cell
- myeloid-derived suppressor cell
- MDSC
1. Introduction
2. Reduced Risk and Progression of HCC in NAFLD/NASH Patients by Metformin
3. Links between NASH and HCC
4. Direct Effects and Underlying Mechanisms of Metformin on Hepatocytes or Malignant Cells that May Inhibit NASH-Related HCC
4.1. Metformin and Substrate Overload in Hepatocyte
Due to its glucose-lowering effect, metformin is likely to relieve the hepatocyte from substrate overload to some extent, which is the stem of NASH pathogenesis, and is associated with the generation of oxidative stress vital in NASH-related HCC development (Figure 2). The glucose-lowering effect of metformin is primarily attributed to its inhibition of hepatocyte gluconeogenesis. However, the underlying mechanism that once seemed to be coined as mediated by the direct inhibition of mitochondrial respiratory complex I by metformin, is still under active debate [94,95]. Despite controversies, metformin is thought to be enriched to mitochondria through the attraction imposed by the mitochondrial membrane potential or through protein-mediated transportation, resulting in a high concentration of the drug and inhibits the mitochondrial respiratory chain, which decreases the ATP/AMP ratio. This altered energy balance in hepatocytes is thought to be the key factor that decreases hepatic gluconeogenesis through multiple pathways. These pathways involve AMP-activated protein kinase (AMPK), fructose 1,6-bisphosphatase (FBP1), adenylate cyclase (AC), acetyl CoA carboxylase (ACC), and others, with many among them having an additional role in NASH pathogenesis and hepatocarcinogenesis. The inhibition of gluconeogenesis and mitochondrial respiratory complex I could be beneficial to NASH-related HCC in several ways. First, inhibition of gluconeogenesis could improve the substrate overload in the hepatocyte, reducing cell stress and postponing the progression of T2DM and NAFLD. It also helps with lowering oncogenic insulin and IGF-1 levels. Inhibition of mitochondrial respiratory complex I by metformin in cancer cells has also been demonstrated, which leads to reduced proliferation, albeit some cancer cell lines are less sensitive due to low dependency on mitochondrial respiration [96,97,98].
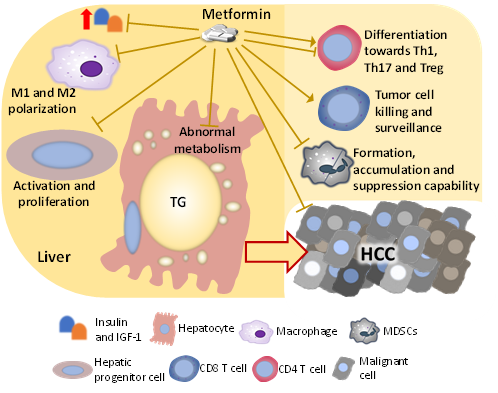
Figure 2. Factors and cells changed by metformin, which mediates the liver-protecting effect against HCC development in the NASH condition. The mechanism by which metformin inhibits the development of NASH-related HCC is multi-factorial. Metformin directly impacts hepatocytes, hepatic progenitor cells, and HCC cells, which suppress malignant transformation and cancer progression. Besides influencing those cells that go through the malignant transformation directly, metformin also changes the activity and population of immune cells including macrophages, T cells, and MDSCs, suppressing the HCC development.
4.2. Metformin and AMPK Signaling
Of those factors regulated by an altered cell energy state induced by metformin, the protein kinase AMPK is highly relevant to NASH-related HCC. Metformin treatment is generally believed to activate AMPK by lowering the cell energy state that promotes the phosphorylation of AMPK by LKB1, even though alternative mechanisms were also proposed [99]. Activated AMPK phosphorylates and inactivates ACCs (ACC1 and ACC2), which are the key enzymes of de novo fatty acid synthesis and the enzyme that produces inhibitors to the fatty acid β-oxidation, respectively [100,101,102]. Thus, the activation of AMPK by metformin can alleviate the substrate overload in hepatocytes by reducing de novo fatty acid synthesis and promoting fatty acid β-oxidation, which could postpone NASH development and progression. Moreover, AMPK signaling has been highlighted in hepatocarcinogenesis and HCC progression, as shown by clinical observations that cirrhosis patients with a low level of AMPK activating phosphorylation having a higher risk for HCC [104], and a low level of AMPK activating phosphorylation in HCC patients with HBV etiology correlating with metastasis and a poor prognosis [11]. Metformin has been shown to suppress the hepatocarcinogenesis of a steatosis-associated mouse liver tumor model of oncogene AKT/c-Met overexpression, where AMPK activation by metformin treatment was seen. Activation of AMPK in cultured human HCC cells by metformin was also shown by the same study [105]. Since the anti-HCC effect and underlying mechanisms of AMPK activation are not specific to NASH-related HCC and have been recently reviewed, this topic will not be discussed here in detail. Still, the mechanisms are generally related to the role of AMPK as a sensor for energy deprivation, where AMPK activation halts the cell cycle and inhibits cell anabolism. The anti-cancer effect of metformin has been attributed to AMPK activation [106]. It is unknown whether NASH-related HCC has more potent or more frequent AMPK inactivation.
4.3. Metformin and Other Factors Regulated by AMP
Metformin has also been shown to inhibit gluconeogenesis by lowering the energy state independent of AMPK activation but by inhibiting FBP1 or AC with increased AMP [109,110]. FBP1 and the product of AC, cAMP, have their role in HCC development. However, inhibition of FBP1 or lowering the cAMP level was shown to be detrimental rather than beneficial in this scenario [111,112,113]. More relevant to NASH-related HCC is that the loss of FBP1 expression induced mild NAFLD-like features in mice and accelerated the progression of the carcinogen-induced liver tumor [112]. In this aspect, using metformin to treat HCC might add fuel to the fire. Further studies that examine these activities of metformin in cancer cells are needed.
4.4. Metformin and Oxidative Stress
Metformin has been demonstrated to alternatively regulate gluconeogenesis by regulating the hepatocyte redox state. It promotes a more reduced cytosol, albeit this effect might be the by-products of the inhibition of the mitochondrial respiratory complex I [94]. The altered hepatocyte redox state by metformin might alleviate oxidative stress, preventing NAFLD progression and hepatocarcinogenesis. Metformin was shown to inhibit oxidative stress-induced apoptosis in primary rat hepatocytes [114]. Metformin was also shown to promote the activation of NRF2, the master regulator of antioxidative response, in the liver of animal models for T2DM or hepatotoxicity [115,116]. In addition, metformin has been shown to activate NRF2 and attenuate oxidative stress independent of AMPK in mouse primary brain endothelial cells [118]. However, the activity of metformin on NRF2 in malignant cells contradicts those shown in non-malignant cells. NRF2 activation in malignant cells helps them to gain survival advantages and mediates drug resistance [119]. Instead of activating NRF2, metformin was shown to induce the downregulation or inactivation of NRF2 in cancer cells [121,122,123,124]. Metformin seems to play opposite roles in terms of NRF2 activation in the malignant and non-malignant cells, which could be owing to different upstream pathways activating NRF2 in the malignant cell as compared to the non-malignant cell, or due to the fact that most studies testing metformin on malignant cells were done in the cell culture system. Despite different effects, current data support that metformin treatment is beneficial in both the NASH and HCC stages in regulating oxidative stress and NRF2 activation.
4.5. Metformin and Hepatic Progenitor Cells
Hepatic progenitor cells (HPCs) are bipotential stem cells that can differentiate into both hepatocyte and cholangiocyte. During chronic liver diseases, including NASH, HPCs are activated and increased to facilitate liver regeneration apart from hepatocytes’ replication [125]. There are different theories, but HPCs can be the origin of HCC cells, and even if HPCs do not directly become cancer cells, they are generally believed to promote carcinogenesis [126]. HPC activation and differentiation are induced by hepatocyte damage and are supported by activated hepatic stellate cells and hepatic macrophages during chronic liver diseases. Thus, hepatocyte-protecting metformin may reduce the number and activation of HPCs to reduce HCC risk in NASH conditions. In a rat model of cirrhosis, metformin was found to reduce HCC incidence by inhibiting HPC activation [127]. A more relevant study was conducted on a unique Ncoa5 deletion mouse model for T2DM and NASH accompanied HCC. Haploid Ncoa5 deficiency induced the appearance of T2DM and NASH features in mice fed a standard diet and caused spontaneous development of HCC, which can be partially attributed to a high expression of pro-inflammatory cytokine IL-6 [128]. In the following study, the group further characterized the oncogenic liver environment and found increased HPC number concurrent with high expression of p21 (p21WAF1/CIP1) in hepatocytes. Metformin was shown to reduce the HCC incidence in this mouse model while reducing p21 expression in hepatocytes and decreasing the HPC number. Deletion of the p21 gene phenocopied metformin treatment in Ncoa5 deficient mice with regard to the reduced HPC number [129]. Thus, metformin may reduce HCC risk in the NASH condition partially by inhibiting HPC activation by reducing p21 expression in hepatocytes. Although metformin has been shown to inhibit p21 expression through AMPK [130], and increased expression of p21 in hepatocytes has been found to increase HPC number 25 years ago [131], it is still not clear how high expression of p21 in hepatocytes promotes HPC activation.
5. Metformin on the Immune Population that May Indirectly Inhibit NASH-Related HCC Development
The immunity in the NASH liver is dysregulated and is generally pro-inflammatory, which stresses and damages the hepatocyte, promoting the accumulation of genetic and epigenetic alterations. Several immunosuppressive components also exist in the dysregulated immunity in the NASH liver, such as M2 macrophages, MDSCs, immunosuppressive B cells, exhausted CD8 T cells, and Tregs, and these components permit the survival and growth of tumor-initiating cells. Metformin has been frequently shown to improve the dysregulated immunity in the liver with chronic diseases including NASH and HCC, which could be partially attributed to the direct hepatocyte-protecting effect, but metformin is also shown to directly act on immune cells (Figure 2).
5.1. Metformin on Macrophages
Suppression of the macrophage activation toward the M1 or M2 phenotype depending on the microenvironment of the specific disease stages could be beneficial to NASH and NASH-related HCC. Inhibiting the M1-related pro-inflammatory activity of macrophages in the early stage of NASH could improve insulin sensitivity [132] and reduce the stress to hepatocytes. At the same time, such inhibition in the tumor or tumor-initiating-cell-bearing liver could be detrimental. Inhibiting the M2-related immuno-modulatory activity of macrophages can remove the permit and support for cancer cell outgrowth. Metformin might inhibit both M1 and M2 phenotypes of macrophages and inhibit the hepatic seeding of macrophages derived from the monocyte in the NASH liver. Moreover, metformin could inhibit M2 polarization of the tumor-associated macrophage in the HCC stage, reinforcing the benefit of metformin use in NASH-related HCC [129,133,134,135,136,137,138,139,140].
5.2. Metformin on MDSCs
Inhibiting the formation and recruitment of MDSCs in the liver would alleviate the immunosuppressive microenvironment that promotes tumor initiation and progression. The accumulation of MDSCs in the NASH liver of Ncoa5+/− mice can be prevented by long-term treatment of metformin, concurrent with a reduced HCC incidence [129]. The reduction of hepatic MDSCs could be the result of suppressed chronic inflammation in the liver by metformin. In the tumor-microenvironment, metformin might also suppress MDSCs’ accumulation. In patients with esophageal squamous cell carcinoma (ESCC), those with diabetes and treated with metformin had significantly less MDSC infiltration in the tumor as compared to those with or without diabetes that were not treated with metformin [141]. The effect was mediated by AMPK activation and subsequent NF-κB inhibition in ESCC cells, and reduced production of chemoattractants for MDSCs. More directly, it has been recently reviewed that the AMPK pathway plays a potential role in regulating MDSC functions, where metformin may inhibit the immunosuppressive function of MDSCs [142]. The AMPK-related inhibition of MDSC functions by metformin was also observed in a syngeneic tumor mouse model [144]. Investigation examining the inhibition of MDSC accumulation and function by metformin in HCC is still lacking, but the discovery of such inhibition in other cancers may apply to the pro-tumorigenic NASH microenvironment and the tumor microenvironment of NASH-related HCC.
5.3. Metformin on T Cells
Inhibiting CD8 T cell infiltration and activation in the early NASH stage can alleviate hepatocyte damage and prevent the activation of immunomodulatory machinery and T cell exhaustion. However, the immunosurveillance by CD8 T cells could be dampened during such an inhibition. In contrast, boosting CD8 T cell expansion and cytotoxicity in the HCC stage can help with tumor clearance. In the NASH liver of the NCOA5-deficient mouse, the increase of hepatic CD8 T cells, which was more likely associated with activated and tissue-resident memory phenotypes, was prevented by long-term metformin treatment concurrent with a reduced enrichment of T cell exhaustion gene signatures in the liver transcriptome [129]. The authors suggested a mechanism related to metformin’s suppression to chronic hepatic inflammation that caused the observed reduction of T cell infiltration and exhaustion. While the study above showed the likely indirect effect of metformin on CD8 T cells in pre-HCC NASH liver, a direct effect of metformin on CD8 T cells was also demonstrated. Murine CD8 T cells treated with metformin more strongly inhibited the tumor growth of a melanoma intradermal inoculation mouse model than non-treated CD8 T cells, and the change depends on AMPK activation by metformin. The same study also showed that oral administration of metformin increased the tumor-infiltration of CD8 T cells and protected them from apoptosis and exhaustion in tumor inoculation mouse models [145]. A similar effect was also seen in the human cell [146]. PD-L1 has recently been found to be phosphorylated by metformin-activated AMPK, and, subsequently, went through ER accumulation and ER-associated protein degradation, leading to enhanced cytotoxicity of T cells [147]. Altogether, it is commonly reported that metformin can boost the anti-tumor activity of CD8 T cells, supporting the use of metformin in NASH-related HCC.
Studies indicated that metformin also regulates the function of CD4 T cells. Direct inhibition of CD4 T cell’s production of IL-22, an HCC-promoting cytokine [148], by metformin has been demonstrated. The effect is mediated by inhibiting the differentiation of CD4 T cells toward Th1 and Th17 cells [149]. The differentiation toward and the function of Treg can be regulated by metformin. In the cell culture system, metformin pre-treatment inhibited the TGF-β-induced CD4 T cell differentiation toward Treg. The resulting Treg with metformin pre-treatment had impaired ability to suppress CD8 T cells. Reduced Treg tumor-infiltration and Treg function by metformin treatment were also found in the intradermal inoculation mouse model of cancer in the same study [150]. It should be noted that, although such inhibition of Treg by metformin can be beneficial in the HCC stage, the immunoregulatory Treg is important in protecting the liver in the NASH stage. In studies related to autoimmune diseases, metformin was shown to promote rather than inhibiting the differentiation toward Treg [151,152]. Metformin may play distinct roles in the differentiation process toward Treg induced by various factors. Still, a consensus, to some extent, has been reached by these studies on metformin’s ability to suppress the differentiation of CD4 T cells toward Th1 and Th17 cells.
6. Conclusions
The prevalence of NAFLD and the rising incidence of NASH-related HCC urge further investigations to understand the mechanism of HCC development with this etiology and find effective drugs to prevent and treat NASH-related HCC. It is supported by accumulating data that metformin can ameliorate NAFLD/NASH-inducing conditions and improve the HCC-inducing features of NASH. Metformin has been shown to act on hepatocytes, HCC cells, and various immune populations to suppress HCC development. Metformin has been recently highlighted in modulating the immunity of the liver against HCC development, and our knowledge is still empty in some areas of this topic. For example, studies examining the effect of metformin on B cells and NK cells in NASH and HCC are still lacking. Clinical studies examining the impact of metformin use in NAFLD/NASH patients on HCC incidence and prognosis start to emerge but are still under-investigated.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22095016