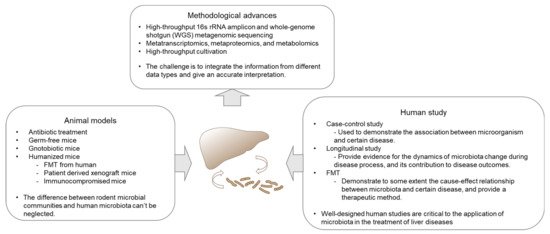
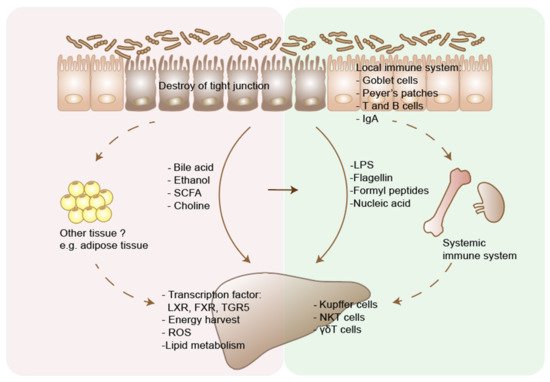
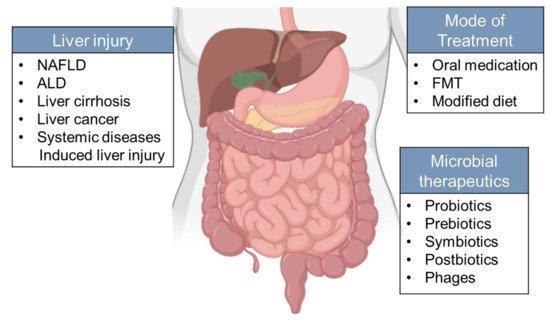
2.1. NAFLD
NAFLD consists of a wide spectrum of liver diseases, ranging from steatosis to nonalcoholic steatohepatitis (NASH), liver fibrosis, and cirrhosis. The pathological process of NASH has been firstly proposed to be the result of “two hits”, which are represented by hepatic steatosis and lipid peroxidation [
18]. This theory later evolved into “multiple hits”, comprising signals derived from the gut or the adipose tissue, such as endotoxin, adiponectin, IL-6, and TNFα [
19]. Besides dietary and genetic factors, the gut microbiota has been demonstrated as an important novel factor in disease pathogenesis, involving in all aspects of the “multiple hits” [
19,
20].
The early disease modality leading to NASH is hepatic steatosis, which is viewed as the liver manifestation of metabolic syndrome. The gut microbiota has been linked to a variety of metabolic disorders, with disease-specific signatures [
21,
22,
23,
24,
25]. Bacterial richness is inversely associated with metabolism-related phenotypes. Individuals with low bacterial richness were at higher odds for insulin resistance, dyslipidemia, and inflammatory phenotype [
21]. A decrease in the proportion of
Bacteroides was observed in obese subjects compared with lean subjects [
26], although this finding is not consistent across all studies [
27]. In addition,
Bacteroides vulgatus was reported to impact the serum metabolome and associate with insulin resistance [
28], and
Bacteroides spp. was associated with hepatic steatosis [
10]. These data suggest versatile roles of gut microbes in different diseases with similar hepatic pathologies.
More than a decade ago, systematic analysis of gut microbiota was conducted in fatty liver disease [
20]. The relation between microbiota and NASH has been discovered in different population-based cohorts. In a pediatric cohort of NASH, Zhu et al. observed a significant increase of the
Enterobacteriaceae family, accompanied with an increase in blood alcohol concentrations, compared to obese and healthy controls [
29]. In view of that
Enterobacteriaceae could produce ethanol toxic to the liver, this evidence proposed a microbiome-based mechanism to explain the histologic similarity between NASH and alcoholic hepatitis. By contrast, in another adolescent cohort of NAFLD and healthy controls, Mouzaki et al. found no statistical difference in
Escherichia coli between two groups [
30]. Besides, they found a decrease of fecal
Bacteroidetes in NASH relative to simple steatosis and healthy controls, which is in agreement with the discoveries in obese patients [
26]. Echoing these findings, an increased abundance of
Bacteroidetes and reduced abundance of
Firmicutes were associated with improvement in hepatic steatosis in a longitudinal study [
31]. However, in a group of biopsy-proven NAFLD patients, Boursier et al. observed a higher abundance of
Bacteroides in NASH and fibrosis patients, compared to those without NASH, suggesting a relation between
Bacteroides and disease severity [
7]. Recently, the high-alcohol-producing
Klebsiella pneumoniae is found to be associated with NAFLD patients in a Chinese cohort [
32]. Again, this data support that an alteration in the gut microbiome contribute to NAFLD through excessing endogenous alcohol production. Other studies, which found no significant difference at the phylum level, documented changes at the class or genus level [
33,
34]. The discrepancies across these studies might result from small numbers of study subject, intrinsic and extrinsic host factors, including age, gender BMI, environmental, geographical, and dietary factors, and even differences in methodology. Despite these variable findings, the correlation of microbiota and NASH has been well recognized.
Recently, the crosstalk between the microbiome and the human host was comprehensively investigated by integrating metagenome, metabolome and hepatic transcriptome and clinical features [
10]. Microbial gene richness decrease was found to be an indicator of deleterious changes. A more complex model was proposed to explain the microbiome–host interplay, involving both metabolism and immune responses. For example, microbial metabolites such as phenylacetic acid (PAA), and microbial products such as Lipopolysaccharide (LPS) have been found to facilitate hepatic lipid accumulation and induce hepatocyte inflammation [
10]. Further investigation revealed that
Proteobacteria,
Actinobacteria and
Verrucomicrobia were positively associated with liver steatosis, whereas
Firmicutes and
Euryarchaeota were negatively associated with liver steatosis [
10]. Overall, the microbial alterations in patients with liver diseases are summarized in .
Harnessing germ-free (GF) animal models, evidence is accumulating to show a causal effect of microbiota in the pathogenesis of metabolic diseases. Germ-free mice were protected against high fat diet (HFD) induced obesity, and conventionalization of GF mice leads to an increase in body-fat content and insulin resistance [
35,
36]. Later analysis found obese microbiome could increase energy harvest from the diet. This trait is transmissible through the colonization of germ-free mice with gut microbiota from obese mice (ob/ob mice) [
37,
38]. Moreover, gut microbiome composition can affect mouse response to HFD, manifested by changes in blood glucose levels and plasma concentrations of pro-inflammatory cytokines. Transplantation of microbiota from HFD mice who have hyperglycemia and higher levels of systemic inflammation can induce the same phenotype in recipient mice [
39]. Fecal microbiome from patients with hepatic steatosis could also trigger steatosis in recipient mice [
10], which further reinforces the significance of microbiota in the pathogenesis of NAFLD.
Table 1. Important studies of human microbiome and liver diseases.
| Study |
Sample Types |
Disease |
Method |
Major Findings |
| Mouzaki et al., 2013, Hepatology [30] |
Stool |
NAFLD |
Quantitative real-time PCR |
The first study assessing the microbiota in adult human NAFLD at different histological stages. Lower percentage of Bacteroidetes is associated with NASH. |
| Jerome et al., 2016, Hepatology [7] |
Stool |
NAFLD |
16s sequencing |
Gut dysbiosis associates with the severity of NAFLD lesions. Bacteroides associated with NASH and Ruminococcus with fibrosis. |
| Loomba et al., 2017, Cell Metabolism [17] |
Stool |
NAFLD |
Shotgun sequencing |
A gut microbiome signature was identified to predict NAFLD with fibrosis. The abundance of Firmicutes and Proteobacteria was higher in mild/moderate fibrosis, and advanced fibrosis, separately. |
| Mutlu et al., 2012, Am J Physiol Gastrointest Liver Physiol [40] |
Human sigmoid mucosa biopsy |
ALD |
Multitag-pyrosequencing |
The first study showing the association between alcohol consumption and microbiome using non-culture method in human. Lower abundances of Bacteroidetes and higher ones of Proteobacteria was found in alcoholics. |
| Dubinkina et al., 2017, Microbiome [9] |
Stool |
ALD |
Shotgun sequencing |
Alcoholic dependence was inversely associated with Clostridiales. The expansion of Bifidobacterium and Lactobacillus was found in ALD patients, but the species were different between patients with and without cirrhosis. |
| Liu et al., 2004, Hepatology [16] |
Stool |
Cirrhosis |
Quantitative bacteriological Culture |
Early evidence of disturbed microecology in cirrhotic patients with MHE, with outgrowth of Escherichia coli and Staphylococcal species. |
| Chen et al., 2011, Hepatology [41] |
Stool |
Cirrhosis |
454 pyrosequencing 16s |
The first major study of microbiome in cirrhosis. Fecal microbial diversity was lower in cirrhotic patients, with higher levels of Enterobacteriaceae and Streptococcaceae, and lower levels of Lachnospiraceae. |
| Bajaj et al. 2014, Journal of Hepatology [42] |
Stool |
Cirrhosis |
Multi-tagged pyrosequencing |
The first major study to compare the microbiome in compensated and decompensated cirrhosis. Introduction of CDR, which is associated with disease progression and prognostic. |
| Qin et al., 2014, Nature [43] |
Stool |
Cirrhosis |
Metagenome sequencing |
The major study characterized the gut microbiome in liver cirrhosis. Patient-enriched species are of buccal origin, with higher levels of Proteobacteria and Fusobacteria and fewer levels of Bacteroides. |
| Caussy C et al., 2019, Nat Commun. [44] |
Stool |
Cirrhosis
(NAFLD-associated) |
16s sequencing |
Bacterial signature was discovered to detect NAFLD-cirrhosis based on a prospective twin and family cohort. |
2.2. ALD
Alcohol has been well demonstrated to be a hepatotoxin, and alcoholic liver disease remains the most prevalent type of chronic liver disease worldwide [
45]. The pathogenesis of ALD includes the toxicity of acetaldehyde, hepatic steatosis, and inflammation. Other factors, both genetic and non-genetic, contribute to interindividual variations in disease development. Among them, the gut microbiota plays a critical role in alcohol-induced liver damage. Alcohol abuse leads to bacterial overgrowth, dysbiosis, and gut barrier dysfunction. The subsequent translocation of bacterial products through portal vein stimulates inflammation and metabolic disorders in the liver [
46].
Dysbiosis refers to the microbial imbalance in the gut. A pioneer study compared the types and numbers of bacteria in the aspirates from the jejunum of patients with and without alcohol abuse and revealed an increase in the number of microorganisms both anaerobic and aerobic, suggesting a role for bacterial overgrowth in the pathogenesis of chronic alcohol abuse [
47]. Later, a study comparing the mucosa-associated colonic microbiome between alcoholics with or without ALD and healthy controls, observed gut microbiome dysbiosis in a subgroup of alcoholics, with lower abundance of
Bacteroidetes and higher ones of
Proteobacteria [
40]. Altered fecal microbiome was also demonstrated in ALD-related cirrhosis compared to healthy subjects [
41,
48].
Murine models of alcoholic liver disease also showed an intestinal bacterial overgrowth and enteric dysbiosis in alcohol fed mice [
49]. Moreover, compared with conventional mice, germ-free mice showed less liver injury after alcohol consumption [
50], corroborating the critical role for gut microbiome in liver disease development. The causal effect of microbiota on ALD has been proposed based on the “leaky gut” theory. The intestinal barrier consists of a mucus layer, physical integrity, and immune defense. Increased intestinal permeability has been described in ALD patients [
51]. Through a chromium-51 absorption test in non-intoxicated alcoholic patients, Bjarnason et al. discovered higher intestinal permeability than controls [
52]. In alcoholics with chronic liver disease, through measurement of urinary excretion of lactulose and mannitol after oral administration, Keshavarzian et al. observed increased intestinal permeability compared to alcoholics with no liver disease and non alcoholics with liver disease [
53]. This “leaky” gut phenotype was also supported by animal models of both acute and chronic ethanol administration [
54]. Acute alcohol intake in rats could increase colonic permeability in 24 h and was associated with significant endotoxemia [
55]. Ten weeks of alcohol gavage also induced gut leakiness in rats, resulting in endotoxemia and liver injury [
56]. Evidence indicates that alcohol-induced gut leakiness and endotoxemia occurs prior to steatohepatitis, acting as a trigger for alcoholic steatohepatitis (ASH) [
57].
Further analysis discovered a negative association between alcohol feeding and the expression of certain bactericidal protein in the host, which can be partially reversed by prebiotics treatment. Other manipulations, such as pectin treatment, and fecal microbiota transplantation (FMT), have been shown to be able to prevent alcohol-induced liver injury in mouse models [
15]. A similar protective function of microbiota has been observed in ALD patients treated with probiotics and fecal transplantation, which leads to improved liver enzyme levels and better clinical outcomes [
58,
59]. Oral supplementation of
Akkermansia muciniphila, which was found to be diminished by ethanol exposure both in mice and humans, can ameliorate experimental ALD and promote intestinal barrier integrity [
60]. However, larger clinical studies across different populations with complete evaluation of benefits versus risks are needed before fecal transplantation or other therapeutic regimes built upon manipulation of the gut microbiota can be considered a routine clinical practice in the treatment of ALD.
2.3. Cirrhosis
Cirrhosis is the end stage of chronic liver disease, which is characterized by the formation of nodules of regenerative parenchyma, often accompanied by portosystemic shunts. Cirrhosis results from different etiologies, covering all the chronic liver diseases mentioned above, with significant differences between regions, genders, and socioeconomic status [
5].
Changes in gut microbiota in patients with liver cirrhosis have been well documented in studies using various approaches. Early studies using bacterial culture found that the microecology was disturbed in cirrhotic patients with minimal hepatic encephalopathy (MHE), characterized by outgrowth of
Escherichia coli and
Staphylococcal species. This can be reversed by synbiotic treatment accompanied by a reduction in blood ammonia and amelioration of MHE [
16]. A later study using 16S rRNA sequencing observed the distinct gut microbiome in liver cirrhosis, showing that at the phylum level,
Bacteroidetes was reduced, while
Proteobacteria and
Fusobacteria were enriched in the fecal microbiota [
41]. Among the bacteria enriched in cirrhosis,
Enterobacteriaceae, Streptococcaceae, and Veillonellaceae were the most abundant families [
41]. A more recent study using quantitative metagenomics revealed that the major change of gut microbiota derives from bloom of oral bacterial species within the gut [
43]. Moreover, a combination of 15 biomarkers was used for discrimination of patients with liver cirrhosis from healthy subjects, highlighting the diagnostic potential of microbial markers for liver cirrhosis [
43]. These microbial genes, with a high specificity to liver cirrhosis, have no overlap with the markers discovered in type 2 diabetes study [
23], which suggests that such microbial features are disease specific.
Other studies have found a correlation between changes of microbiota and severity of disease. Bajaj et al. established a cirrhosis dysbiosis ratio (CDR) based on the ratio of autochthonous to pathogenic taxa, with a low CDR associated with worse disease state [
42]. In a longitudinal analysis, comparing patients before and after hepatic encephalopathy (HE) development, CDR was also found to be decreased in patients after occurrence of HE [
42]. Loomba et al. showed a shift in microbiome composition during disease progression from mild NAFLD to advanced fibrosis, with an increase in
Proteobacteria and a decrease in
Firmicutes at the phylum level. At the species level,
E. rectale and
B. vulgatus were the most abundant microorganisms in mild and advanced fibrosis, respectively [
17]. They also identified 37 microbial species which distinguish different stages of the disease, suggesting the potential use of microbial markers as a tool in diagnosing and determining the stages of liver disease. Qin et al. found that the severity of liver cirrhosis correlated with the abundance of orally derived species in the gut [
43]. In line with these results, studies trying to investigate the effect of gut bacteria on the gut-liver-brain axis by magnetic resonance imaging (MRI), found a positive correlation of
Enterobacteriaceae and
Streptococcacae with astrocytic changes. In addition,
Porphyromonadaceae is associated with changes of neuronal integrity and edema [
61].
Commonly used liver cirrhosis/fibrosis mouse models include chemical-based models, which is induced by carbon tetrachloride, thioacetamide or ethanol, diet-based models, mostly methionine-deficient and choline-deficient diet, and surgery-based models, as the common bile duct ligation [
62]. Although differences exist between the cause of liver cirrhosis in human and in mouse models, significant change in the gut microbiome were observed in nearly each model [
63]. The underlying mechanism of how microbiota effect liver cirrhosis has also been explored by using animal models, which include regulation of gut permeability to release bacterial products and regulation of liver metabolism [
64]. However, with the large differences between intestinal microbiota in mice and humans [
65], to translate the knowledge gained from mouse studies to human should be very careful [
66].
2.4. Liver Cancer
The development of liver cancer is a multi-step process, involving factors including genetics, environmental factors, metabolism, and the immune system. Alterations in the microbiota have been reported to contribute to the development of cancer and modulate the efficacy of cancer therapy [
67].
In a human study of cirrhotic patients with HCC, the fecal microbiota dysbiosis is characterized by an overgrowth of
E. coli [
68]. In human liver samples, helicobacter was detected in liver carcinoma, while no evidence of helicobacter can be found in patients without malignancy [
69]. Studies in animal models have shown that a lack of gut bacteria prevented development of hepatocellular carcinoma (HCC) in different models [
6,
70]. In HCC mouse model using DEN (diethylnitrosamine) plus CCl
4 treatment, LPS from the intestinal microbiota contributes to tumor growth by promoting liver cell proliferation, suggesting a role for gram-negative bacteria in tumorgenesis [
6]. On the other hand, gram-positive bacterial strains were shown to be increased in mice fed with HFD, and deletion of gram-positive bacteria with vancomycin alleviated the development of HCC induced by DMBA (7,12-dimethylbenz(a) anthracene) and HFD [
70].
In addition to the accumulating evidence suggestive of the relation between microbiota and cancer development, the potential role of microbiota in anticancer therapy has been studied [
71,
72]. In melanoma patients, higher gut microbiome diversity and a relative higher abundance of
Ruminococcaceae are related with better response to anti-PD-1 immunotherapy [
71]. Further study by transplantation (FMT) of stool from responders to anti-PD-1 therapy to germ-free mice showed reduced tumor growth and more importantly, improved responses to anti-PD-1 therapy [
71]. In tumor-bearing mice, gut microbiota has been shown to shape the antitumor immune response through translocation into secondary lymphoid organs and thereby stimulating T helper cells [
72]. However, no study directly tested the effect of modulating gut microbiota in the treatment of HCC. Targeting the gut–microbiota–liver axis represents an attractive therapeutic option for HCC treatment [
73].