Nanobodies are highly water-soluble and stable, have high specificity, and can bind their targets with very high affinity, often in the low nanomolar range.
- nanobodies
- VHHs
- molecular imaging
- radionuclide imaging
- immuno-PET
- SPECT/CT
- cancer-specific markers
- immune checkpoint imaging
1. Introduction
In addition to conventional antibodies, camelids, such as llamas and alpacas, have unique heavy-chain-only antibodies [1]. These antibodies are unique in that the variable regions are encompassed by a single domain (VHH) instead of two separate domains (VH and VL) as seen in conventional antibodies [2]. The variable domains of the camelid heavy-chain-only antibodies have found widespread applications in biomedical research.
Nanobodies are highly water-soluble and stable, have high specificity, and can bind to their targets with high affinity, often in the low nanomolar range [3]. VHHs are stable as single-domain antibodies because of several mutations on their surface that allow them to be water soluble [3]. In particular, several residues that would be at the VH–VL interface in conventional antibodies are mutated for hydrophobic to hydrophilic residues (G44E, L45R, and W47G) (Figure 1), enhancing their stability and solubility as a single domain. In addition, there is a solubility enhancing mutation, most commonly found in camel VHHs, at the VH–CH1 interface (L11S) (Figure 1A,C).
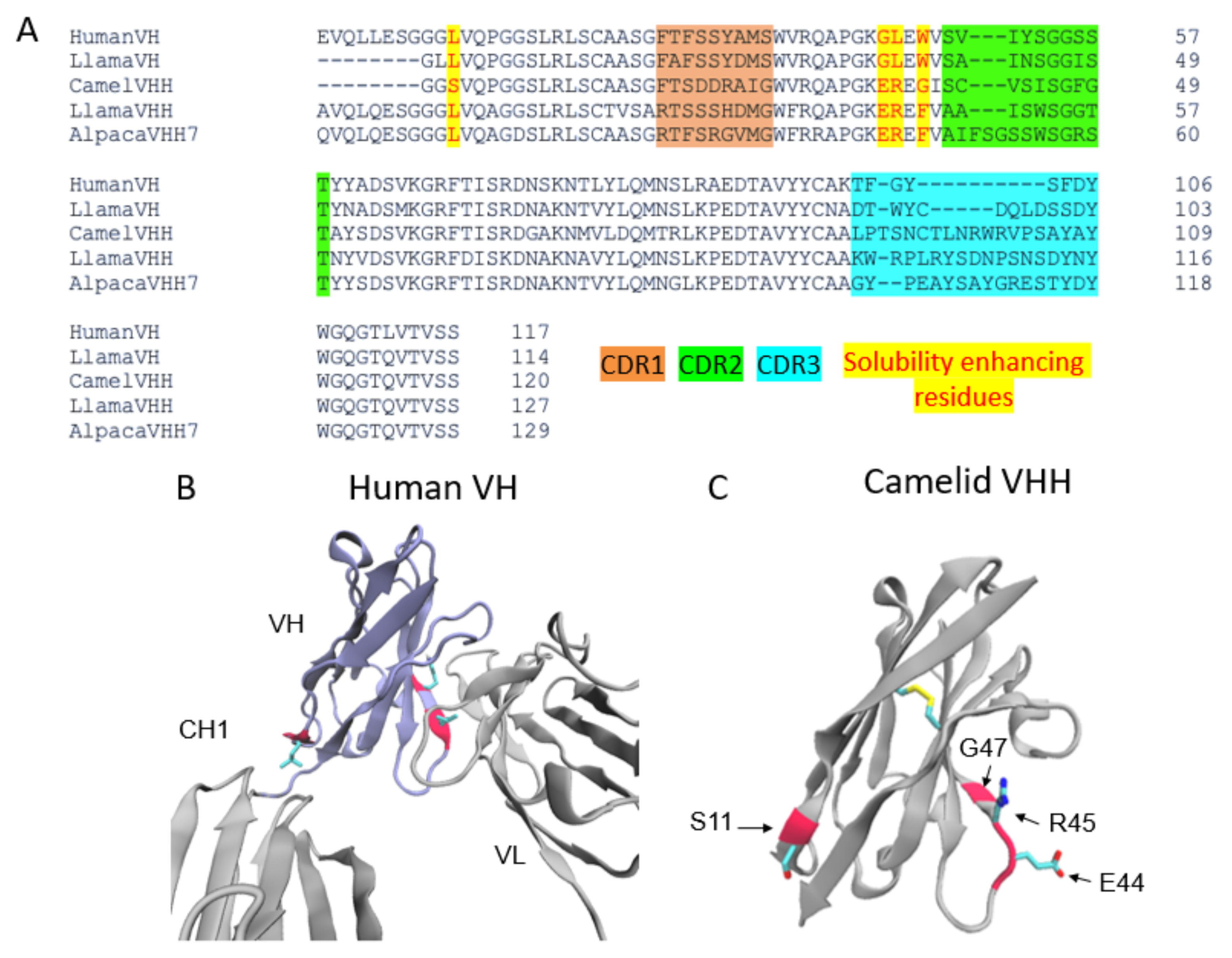
Figure 1. The architecture of VHHs promotes their solubility and stability. (A) Multiple sequence alignment of the Human VH, Llama VH, Llama VHH, Camel VHH, and Alpaca VHH domains. CDR regions are highlighted orange (CDR1), green (CDR2), and blue (CDR3). The VHH domains shown have a larger CDR3 region than the VH CDR3s. Highlighted in yellow and written in red are residues mutated to enhance solubility in VHH domains. Camel VHH bears four mutations (L11S, G44E, L45R, and W47G). Llama and alpaca VHHs bear G44E, L45R, as well as a W47F mutation. (B) Crystal structure (PDB ID 1IGY) of a human antibody with the VH domain colored in purple. Residues mutated in the camelid VHH domains are colored red. L11 makes contact with the CH1 domain while L45 and W47 form contacts with the VL domain. (C) Crystal structure of a camelid (camel) VHH (PDB ID 6U14). E44, R45, and G47 in the hypothetical VL binding site promote stability and solubility of the VHH as a single domain.
The factor contributing to the high affinity of these nanobodies is that their frameworks have three complementarity-determining regions (CDRs). These CDRs are analogous to those found in human antibody VH and VL domains and are subject to somatic hypermutation in the course of affinity maturation. The CDR3 of VHHs is especially long in comparison to the human counterpart [4]. The length and flexibility of VHH CDR3s enable the nanobody to access a variety of conformations. In some cases, VHH CDR3s are able to fold back and make contact with the nanobody framework [4]. Taken together, these factors compensate for the lack of sequence variability incurred by the loss of VL CDRs, allowing VHHs to bind to their targets with high specificity and affinity (Figure 1A).
Methods of generating nanobodies against an antigen of interest have already been well established [2]. In brief, a llama or alpaca (among a variety of other camelids) is immunized against the antigen(s) of interest [2]. Administration of the protein antigen is typically accompanied by an immune adjuvant that serves to enhance the overall immune response [5]. Several weeks later, blood is harvested from the immunized animal and peripheral blood mononuclear cells (PBMCs) are purified. This purification is then followed by total RNA extraction, VHH amplification, and finally, the construction of a phage display library. Phage display libraries are among the most common methods of preparing nanobody libraries, but other methods, such as E. coli or yeast display, could alternatively be used [2][6][7]. Finally, the lead VHHs are identified and expressed as soluble proteins using reliable approaches, such as magnetic-activated cell sorting (MACS), fluorescence-activated cell sorting (FACS), or panning against immobilized antigens (Figure 2) [8][9][10].
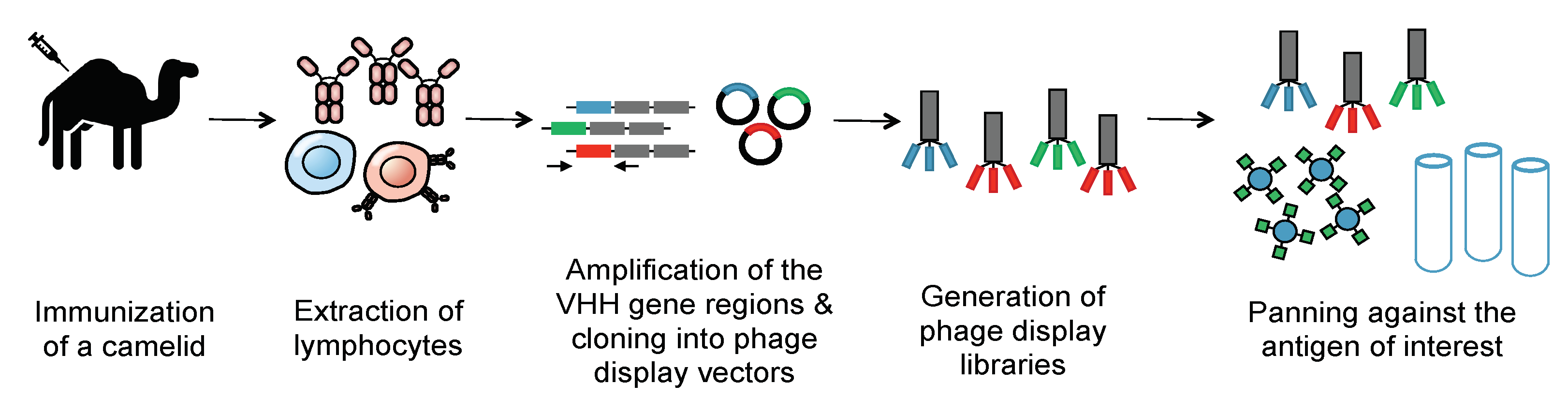
Figure 2. Generation of a nanobody library. To create an immune library, camelids are immunized against a molecule of interest. mRNA of the camelids’ peripheral blood mononuclear cells is then converted into cDNA. PCR is then employed to amplify the VHH genes. These immune VHH genes will then be cloned into a phage display vector. Phages are then generated using E. coli strains such as TG1. Phage libraries are then panned against immobilized antigens to select for nanobodies that selectively bind the antigen with high affinity. The panned libraries are then used for reinfection of E. coli to obtain specific clones.
The short circulatory half-life of nanobodies have allowed the use of a range of isotopes with short half-lives for imaging, such as Galium-68 (68Ga, t1/2 = 67.71 min) and 18F (t1/2 = 109.7 min), as well as longer-lived isotopes, such as Technetium-99m (99mTc t1/2 = 6.0 h), Copper-64 (64Cu t1/2 = 12.7 h), Indium-111 (111In t1/2 = 67.2 h), Zirconium-89 (89Zr t1/2 = 78.41 h), and Lutetium-177 (177Lu t1/2 = 6.7 days). Similar to other antibody fragments, nanobodies are commonly labeled nonspecifically via their side-chain lysine residues using chelators or radioisotopes that are functionalized with amine-reactive groups such as N-hydroxysuccinimide (NHS) or isothiocyanatobenzyl (pSCN) groups. While this strategy is robust and reproducible, it is not site-specific, which may damage antigen-binding sites [11]. To address this issue and to ensure the binding capacity is not compromised, a variety of site-specific labeling approaches, such as the use of sortase technology, have been developed [12]. Another common approach is using a His6 tag to install 99mTc, a commonly used SPECT isotope [13].
2. Nanobodies for Medical Imaging
Biopsies will likely remain the gold standard of cancer diagnostics for the foreseeable future; however, biopsies can sometimes be unrepresentative of the greater TME or targeted organ. Non-invasive immuno-PET imaging, as an adjunct to biopsies, can provide a holistic view of the TME and offer a complete insight into both primary and metastatic tumors. Information revealed via imaging can help to make informed treatment decisions. Imaging is also beneficial in understanding the progression and pathogenesis of a variety of diseases, such as fibrosis, cardiovascular complications, arthritis, and neurological diseases. Therefore, immuno-PET imaging is a potentially revolutionary addition to disease management and treatment.
The use of radiolabeled nanobodies as imaging probes overcomes many of the weaknesses of using full-size antibodies and larger antibody fragments. Nanobodies have excellent tissue penetration, rapid blood clearance profile, high specificity, low nanomolar to picomolar affinity for the target, high stability and water solubility, ease of production, and a pharmacokinetic profile compatible with short-lived radioisotopes. One major drawback of nanobody-PET is the high renal retention and toxicities, similar to other radiotherapeutics and imaging agents [14][15]; however, techniques such as PEGylation can help decrease kidney retention [12]. Other strategies to decrease renal toxicity involve the co-infusion of basic amino acids such as lysines, or the use of gelofusine, which is a gelatin-based plasma expander [16][17]. Gelofusine mediates a decrease in kidney uptake through the interference of its plasma expander with the tubular reabsorption of nanobodies. Immunogenicity has also been reported for some nanobodies, though it may be idiotypic and specific only to the variable regions. Once again, techniques such as PEGylation may help to decrease immunogenicity in addition to undesirable kidney retention [1][18]. A recent study assessed the immunogenicity risk profiles of two nanobodies –– anti-HER2 and anti-CD206 (MMR) –– that have advanced into Phase II clinical trials for PET imaging. Strikingly, only 1 patient out of 20 showed a minimum amount of pre-existing anti-VHH antibodies, which was only marginally increased several months post-injection of the nanobody. Assessing the in vitro immunogenicity of the nanobodies using human dendritic cells did not induce T cell activation, further suggesting a low immunogenicity profile of nanobodies [19].
These issues of kidney retention and immunogenicity require better understanding and further investigation, as overcoming them would contribute greatly to clinical success. Similar to other antibody-based imaging approaches, nanobody-based imaging agents are the most effective when selected epitopes demonstrate a few common characteristics. These include antigen recognition through expression on the extracellular surface of the plasma membrane, availability of the epitope for similar recognition, high expression of the antigen on the cell surface, and little to no expression in normal tissues.
Radiolabeled nanobodies can provide valuable information about the biological processes taking place inside living organisms, giving researchers and clinicians the appropriate data that are needed to improve patient care. For example, nanobodies can be used to understand the dynamic of immune responses, helping to gain mechanistic insight into how the tumor immune landscape is shaped and responds to treatment. Understanding the response mechanisms, in turn, may lead to the identification of new targets and avenues to pursue for developing new therapeutics or biomarkers. Several studies have been performed on imaging lymphocytes, checkpoint molecules, and cancer markers; the recent more in-depth understanding of the tumor immune landscape suggests myeloid cells play a central role in shaping the TME. Therefore, pursuing the development of novel nanobodies for imaging specific subsets of myeloid cells can turn out to be both important and advantageous. Similarly, as cytokines and chemokines are key players in the pathogenesis of the disease, imaging their level of presence and movement may help to gain insight into understudied disease pathogeneses and progression models. Taken together, understanding the behavior of immune cells and immune-modulating molecules before, during, and after treatment will help to decide the best course of treatment for patients and allow for a dynamic and adaptable inpatient care experience. Harnessing the powerful imaging potential of nanobodies would be an ideal strategy to tackle these issues and ultimately achieve such ideal outcomes.
To ensure maximal effectiveness and minimal nonspecific binding, it could be worthwhile to continue searching for other targetable markers associated with the diseases mentioned in this review, but the major priority in coming years may better be focused to expand the number of different diseases that can be imaged and characterized by radiolabeled nanobodies, such as inflammation markers to diagnose fever of unknown origin, neurodegenerative diseases such as Alzheimer, and cytokines, chemokines, and their receptors that are key in pathogenesis and progression of the disease. By helping scientists better understand and visualize the driving forces behind disease progression, expanding the library of nanobody-based imaging agents will become an even more promising tool in guiding the development of novel, effective treatment plans for patients in the future.
The generation of nanobodies is now a well-established procedure [2][20]. The increasing availability of commercial sources for immunization and identification of lead candidates, along with advancements in the development of synthetic libraries will continue to help provide easier access to new nanobodies against antigens of interest. While we have focused on the imaging applications of nanobodies, they also can be used as therapeutics, as a molecular biology tool for mechanistic studies, and to investigate biological processes. With the recent FDA approval of a nanobody-based treatment (Caplacizumab, a bivalent nanobody) and the clinical translation of several nanobodies, the repertoire of available nanobodies is only expected to grow in the years to come (Table 1).
Table 1. Nanobodies developed for noninvasive immuno-PET/SPECT imaging.
| Target | Agent | Reactivity | Clinical Trials: Stage and Status (If Applicable) | References |
|---|---|---|---|---|
| EGFR | 99mTc-8B6 | Human | Preclinical | [7] |
| 99mTc-7C12 | Human | Preclinical | [21] | |
| HER2 | 177Lu-2Rs15dHIS | Human | Preclinical | [22] |
| 18F-FB-2Rs15d | Murine | Preclinical | [23] | |
| 18F-RL-I-5F7 | Murine | Preclinical | [24] | |
| 68Ga-2Rs15d | Human | Clinical | [23][25] | |
| HER3 | 89Zr-MSB0010853 | Murine | Preclinical | [26] |
| CEA | 99mTc-NbCEA5 | Human | Preclinical | [27] |
| PSMA | 111In-JVZ007 | Human | Preclinical | [28] |
| HGF | 89Zr-1E2, 89Zr-6E10 | Human | Preclinical | [29] |
| CD20 | 68Ga-9079 | Human | Preclinical | [30] |
| CD38 | 68Ga-NOTA-Nb1053 | Murine | Preclinical | [31] |
| Mesothelin | 99mTc-A1, 99mTc-C6 | Human | Preclinical | [32] |
| MMR | 99mTc-d a-MMR Nb cl1 | Murine | Preclinical | [33][34] |
| 18 F-FB-anti-MMR 3.49 | Human, Murine | Preclinical | [35] | |
| 68Ga-NOTA-Anti-MMR-VHH2 | Human | Clinical, NCT04168528 (Active) | [36] | |
| MHC II | [18F]FDG -VHH7 | Murine | Preclinical | [37] |
| 64Cu- VHH4 | Human | Preclinical | [38] | |
| CD11b | 89Zr-VHHDC13 (PEGylated) |
Murine | Preclinical | [39] |
| 18F-VHHDC13 | Human | Preclinical | [40] | |
| CD8 | 89Zr-VHH-X118 (PEGylated) |
Murine | Preclinical | [12] |
| 68Ga-NOTA-SNA006 | Human | Preclinical | [41] | |
| Mouse Dendritic Cells | 99mTc-Nb-DC2.1 | Murine | Preclinical | [42] |
| 99mTc-Nb-DC1.8 | Murine | Preclinical | [42] | |
| PD-L1 | 18F-B3, 18F-A12, 64Cu-B3 | Murine | Preclinical | [43] |
| 99mTc-C3, 99mTc-C7, 99mTc-E2, 99mTc-E4, 99mTc-K2 | Murine | Preclinical | [44][45][46][47] | |
| 68Ga-NOTA-Nb109 | Human | Preclinical | [48] | |
| 99mTc-NM-01 | Human | Clinical, NCT02978196 (Concluded) |
[49] | |
| 89Zr-envafolimab (Fc fusion) |
Human | Clinical, NCT03638804 (Active) |
[50][51] | |
| CTLA-4 | 18F-H11, 89Zr-H11 | Murine | Preclinical | [51][52] |
| LAG-3 | 99mTc-anti-moLAG-3 3206, 99mTc-anti-moLAG-3 3208, 99mTc-anti-moLAG-3 3132, 99mTc-anti-moLAG-3 3141 |
Murine | Preclinical | [53][54] |
| VCAM-1 | 99m Tc-cAbVCAM1-5 | Human, Murine | Preclinical | [33][55][56][57] |
| FN-EIIIB (ECM) | 64 Cu-NJB2 | Human, Murine | Preclinical | [58] |
| αSyn | NbSyn2, NbSyn87 (fused to fluorescent proteins for imaging) | Human | Preclinical | [59][60] |
| DPP6 | 99m Tc-4hD29 | Human | Preclinical | [61] |
| Vsig4 | 99m Tc-NbV4 | Murine | Preclinical | [62][63] |
| Clec4F (KC) | 99m Tc-NbC4 | Murine | Preclinical | [62] |
This entry is adapted from the peer-reviewed paper 10.3390/biom11050637
References
- Bannas, P.; Hambach, J.; Koch-Nolte, F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front. Immunol. 2017, 8, 1603.
- Pardon, E.; Laeremans, T.; Triest, S.; Rasmussen, S.G.F.; Wohlkönig, A.; Ruf, A.; Muyldermans, S.; Hol, W.G.J.; Kobilka, B.K.; Steyaert, J. A General Protocol for the Generation of Nanobodies for Structural Biology. Nat. Protoc. 2014, 9, 674–693.
- Debie, P.; Devoogdt, N.; Hernot, S. Targeted Nanobody-Based Molecular Tracers for Nuclear Imaging and Image-Guided Surgery. Antibodies 2019, 8, 12.
- Mitchell, L.S.; Colwell, L.J. Comparative Analysis of Nanobody Sequence and Structure Data. Proteins Struct. Funct. Bioinform. 2018, 86, 697–706.
- Muyldermans, S. A Guide to: Generation and Design of Nanobodies. FEBS J. 2020.
- Fleetwood, F.; Devoogdt, N.; Pellis, M.; Wernery, U.; Muyldermans, S.; Ståhl, S.; Löfblom, J. Surface Display of a Single-Domain Antibody Library on Gram-Positive Bacteria. Cell. Mol. Life Sci. CMLS 2013, 70, 1081–1093.
- Koide, A.; Koide, S. Affinity Maturation of Single-Domain Antibodies by Yeast Surface Display. Methods Mol. Biol. Clifton NJ 2012, 911, 431–443.
- Salema, V.; Fernández, L.Á. Escherichia Coli Surface Display for the Selection of Nanobodies. Microb. Biotechnol. 2017, 10, 1468–1484.
- Salema, V.; Mañas, C.; Cerdán, L.; Piñero-Lambea, C.; Marín, E.; Roovers, R.C.; Van Bergen En Henegouwen, P.M.P.; Fernández, L.Á. High Affinity Nanobodies against Human Epidermal Growth Factor Receptor Selected on Cells by E. coli Display. mAbs 2016, 8, 1286–1301.
- McMahon, C.; Baier, A.S.; Pascolutti, R.; Wegrecki, M.; Zheng, S.; Ong, J.X.; Erlandson, S.C.; Hilger, D.; Rasmussen, S.G.F.; Ring, A.M.; et al. Yeast Surface Display Platform for Rapid Discovery of Conformationally Selective Nanobodies. Nat. Struct. Mol. Biol. 2018, 25, 289–296.
- Higashikawa, K.; Yagi, K.; Watanabe, K.; Kamino, S.; Ueda, M.; Hiromura, M.; Enomoto, S. 64Cu-DOTA-Anti-CTLA-4 MAb Enabled PET Visualization of CTLA-4 on the T-Cell Infiltrating Tumor Tissues. PLoS ONE 2014, 9, e109866.
- Rashidian, M.; Ingram, J.R.; Dougan, M.; Dongre, A.; Whang, K.A.; LeGall, C.; Cragnolini, J.J.; Bierie, B.; Gostissa, M.; Gorman, J.; et al. Predicting the Response to CTLA-4 Blockade by Longitudinal Noninvasive Monitoring of CD8 T Cells. J. Exp. Med. 2017, 214, 2243–2255.
- Xavier, C.; Devoogdt, N.; Hernot, S.; Vaneycken, I.; D’Huyvetter, M.; De Vos, J.; Massa, S.; Lahoutte, T.; Caveliers, V. Site-Specific Labeling of His-Tagged Nanobodies with 99mTc: A Practical Guide. Methods Mol. Biol. Clifton NJ 2012, 911, 485–490.
- Lambert, B.; Cybulla, M.; Weiner, S.M.; Van De Wiele, C.; Ham, H.; Dierckx, R.A.; Otte, A. Renal Toxicity after Radionuclide Therapy. Radiat. Res. 2004, 161, 607–611.
- Vegt, E.; de Jong, M.; Wetzels, J.F.M.; Masereeuw, R.; Melis, M.; Oyen, W.J.G.; Gotthardt, M.; Boerman, O.C. Renal Toxicity of Radiolabeled Peptides and Antibody Fragments: Mechanisms, Impact on Radionuclide Therapy, and Strategies for Prevention. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2010, 51, 1049–1058.
- van Eerd, J.E.M.; Vegt, E.; Wetzels, J.F.M.; Russel, F.G.M.; Masereeuw, R.; Corstens, F.H.M.; Oyen, W.J.G.; Boerman, O.C. Gelatin-Based Plasma Expander Effectively Reduces Renal Uptake of 111In-Octreotide in Mice and Rats. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2006, 47, 528–533.
- Vegt, E.; Wetzels, J.F.M.; Russel, F.G.M.; Masereeuw, R.; Boerman, O.C.; van Eerd, J.E.; Corstens, F.H.M.; Oyen, W.J.G. Renal Uptake of Radiolabeled Octreotide in Human Subjects Is Efficiently Inhibited by Succinylated Gelatin. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2006, 47, 432–436.
- Jovčevska, I.; Muyldermans, S. The Therapeutic Potential of Nanobodies. BioDrugs 2020, 34, 11–26.
- Ackaert, C.; Smiejkowska, N.; Xavier, C.; Sterckx, Y.G.J.; Denies, S.; Stijlemans, B.; Elkrim, Y.; Devoogdt, N.; Caveliers, V.; Lahoutte, T.; et al. Immunogenicity Risk Profile of Nanobodies. Front. Immunol. 2021, 12, 632687.
- Zimmermann, I.; Egloff, P.; Hutter, C.A.J.; Kuhn, B.T.; Bräuer, P.; Newstead, S.; Dawson, R.J.P.; Geertsma, E.R.; Seeger, M.A. Generation of Synthetic Nanobodies against Delicate Proteins. Nat. Protoc. 2020, 15, 1707–1741.
- Gainkam, L.O.T.; Keyaerts, M.; Caveliers, V.; Devoogdt, N.; Vanhove, C.; Van Grunsven, L.; Muyldermans, S.; Lahoutte, T. Correlation between Epidermal Growth Factor Receptor-Specific Nanobody Uptake and Tumor Burden: A Tool for Noninvasive Monitoring of Tumor Response to Therapy. Mol. Imaging Biol. 2011, 13, 940–948.
- D’Huyvetter, M.; Aerts, A.; Xavier, C.; Vaneycken, I.; Devoogdt, N.; Gijs, M.; Impens, N.; Baatout, S.; Ponsard, B.; Muyldermans, S.; et al. Development of 177Lu-Nanobodies for Radioimmunotherapy of HER2-Positive Breast Cancer: Evaluation of Different Bifunctional Chelators: 177LU-NANOBODIES FOR RADIOIMMUNOTHERAPY. Contrast Media Mol. Imaging 2012, 7, 254–264.
- Xavier, C.; Blykers, A.; Vaneycken, I.; D’Huyvetter, M.; Heemskerk, J.; Lahoutte, T.; Devoogdt, N.; Caveliers, V. (18)F-Nanobody for PET Imaging of HER2 Overexpressing Tumors. Nucl. Med. Biol. 2016, 43, 247–252.
- Vaidyanathan, G.; McDougald, D.; Choi, J.; Koumarianou, E.; Weitzel, D.; Osada, T.; Lyerly, H.K.; Zalutsky, M.R. Preclinical Evaluation of 18F-Labeled Anti-HER2 Nanobody Conjugates for Imaging HER2 Receptor Expression by Immuno-PET. J. Nucl. Med. 2016, 57, 967–973.
- Zhou, Z.; Vaidyanathan, G.; McDougald, D.; Kang, C.M.; Balyasnikova, I.; Devoogdt, N.; Ta, A.N.; McNaughton, B.R.; Zalutsky, M.R. Fluorine-18 Labeling of the HER2-Targeting Single-Domain Antibody 2Rs15d Using a Residualizing Label and Preclinical Evaluation. Mol. Imaging Biol. 2017, 19, 867–877.
- Warnders, F.J.; Terwisscha van Scheltinga, A.G.T.; Knuehl, C.; van Roy, M.; de Vries, E.F.J.; Kosterink, J.G.W.; de Vries, E.G.E.; Lub-de Hooge, M.N. Human Epidermal Growth Factor Receptor 3-Specific Tumor Uptake and Biodistribution of 89Zr-MSB0010853 Visualized by Real-Time and Noninvasive PET Imaging. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2017, 58, 1210–1215.
- Vaneycken, I.; Govaert, J.; Vincke, C.; Caveliers, V.; Lahoutte, T.; De Baetselier, P.; Raes, G.; Bossuyt, A.; Muyldermans, S.; Devoogdt, N. In Vitro Analysis and in Vivo Tumor Targeting of a Humanized, Grafted Nanobody in Mice Using Pinhole SPECT/Micro-CT. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2010, 51, 1099–1106.
- Chatalic, K.L.S.; Veldhoven-Zweistra, J.; Bolkestein, M.; Hoeben, S.; Koning, G.A.; Boerman, O.C.; de Jong, M.; van Weerden, W.M. A Novel 111In-Labeled Anti-Prostate-Specific Membrane Antigen Nanobody for Targeted SPECT/CT Imaging of Prostate Cancer. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2015, 56, 1094–1099.
- Vosjan, M.J.W.D.; Vercammen, J.; Kolkman, J.A.; Stigter-van Walsum, M.; Revets, H.; van Dongen, G.A.M.S. Nanobodies Targeting the Hepatocyte Growth Factor: Potential New Drugs for Molecular Cancer Therapy. Mol. Cancer Ther. 2012, 11, 1017–1025.
- Krasniqi, A.; D’Huyvetter, M.; Xavier, C.; Van der Jeught, K.; Muyldermans, S.; Van Der Heyden, J.; Lahoutte, T.; Tavernier, J.; Devoogdt, N. Theranostic Radiolabeled Anti-CD20 SdAb for Targeted Radionuclide Therapy of Non-Hodgkin Lymphoma. Mol. Cancer Ther. 2017, 16, 2828–2839.
- Wang, C.; Chen, Y.; Hou, Y.N.; Liu, Q.; Zhang, D.; Zhao, H.; Zhang, Y.; An, S.; Li, L.; Hou, J.; et al. ImmunoPET Imaging of Multiple Myeloma with [68Ga]Ga-NOTA-Nb1053. Eur. J. Nucl. Med. Mol. Imaging 2021.
- Montemagno, C.; Bacot, S.; Ahmadi, M.; Kerfelec, B.; Baty, D.; Debiossat, M.; Soubies, A.; Perret, P.; Riou, L.; Fagret, D.; et al. Preclinical Evaluation of Mesothelin-Specific Ligands for SPECT Imaging of Triple-Negative Breast Cancer. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2018, 59, 1056–1062.
- Put, S.; Schoonooghe, S.; Devoogdt, N.; Schurgers, E.; Avau, A.; Mitera, T.; D’Huyvetter, M.; De Baetselier, P.; Raes, G.; Lahoutte, T.; et al. SPECT Imaging of Joint Inflammation with Nanobodies Targeting the Macrophage Mannose Receptor in a Mouse Model for Rheumatoid Arthritis. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2013, 54, 807–814.
- Movahedi, K.; Schoonooghe, S.; Laoui, D.; Houbracken, I.; Waelput, W.; Breckpot, K.; Bouwens, L.; Lahoutte, T.; Baetselier, P.D.; Raes, G.; et al. Nanobody-Based Targeting of the Macrophage Mannose Receptor for Effective In Vivo Imaging of Tumor-Associated Macrophages. Cancer Res. 2012, 72, 4165–4177.
- Blykers, A.; Schoonooghe, S.; Xavier, C.; D’hoe, K.; Laoui, D.; D’Huyvetter, M.; Vaneycken, I.; Cleeren, F.; Bormans, G.; Heemskerk, J.; et al. PET Imaging of Macrophage Mannose Receptor-Expressing Macrophages in Tumor Stroma Using 18F-Radiolabeled Camelid Single-Domain Antibody Fragments. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2015, 56, 1265–1271.
- Xavier, C.; Blykers, A.; Laoui, D.; Bolli, E.; Vaneyken, I.; Bridoux, J.; Baudhuin, H.; Raes, G.; Everaert, H.; Movahedi, K.; et al. Clinical Translation of [68Ga]Ga-NOTA-Anti-MMR-SdAb for PET/CT Imaging of Protumorigenic Macrophages. Mol. Imaging Biol. 2019, 21, 898–906.
- Rashidian, M.; Keliher, E.; Dougan, M.; Juras, P.K.; Cavallari, M.; Wojtkiewicz, G.R.; Jacobsen, J.; Edens, J.G.; Tas, J.M.G.; Victora, G.; et al. The Use of (18)F-2-Fluorodeoxyglucose (FDG) to Label Antibody Fragments for Immuno-PET of Pancreatic Cancer. ACS Cent. Sci. 2015, 1, 142–147.
- Van Elssen, C.H.M.J.; Rashidian, M.; Vrbanac, V.; Wucherpfennig, K.W.; Habre, Z.E.; Sticht, J.; Freund, C.; Jacobsen, J.T.; Cragnolini, J.; Ingram, J.; et al. Noninvasive Imaging of Human Immune Responses in a Human Xenograft Model of Graft-Versus-Host Disease. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2017, 58, 1003–1008.
- Rashidian, M.; LaFleur, M.W.; Verschoor, V.L.; Dongre, A.; Zhang, Y.; Nguyen, T.H.; Kolifrath, S.; Aref, A.R.; Lau, C.J.; Paweletz, C.P.; et al. Immuno-PET Identifies the Myeloid Compartment as a Key Contributor to the Outcome of the Antitumor Response under PD-1 Blockade. Proc. Natl. Acad. Sci. USA 2019, 116, 16971–16980.
- Rashidian, M.; Keliher, E.J.; Bilate, A.M.; Duarte, J.N.; Wojtkiewicz, G.R.; Jacobsen, J.T.; Cragnolini, J.; Swee, L.K.; Victora, G.D.; Weissleder, R.; et al. Noninvasive Imaging of Immune Responses. Proc. Natl. Acad. Sci. USA 2015, 112, 6146–6151.
- Zhao, H.; Wang, C.; Yang, Y.; Sun, Y.; Wei, W.; Wang, C.; Wan, L.; Zhu, C.; Li, L.; Huang, G.; et al. ImmunoPET Imaging of Human CD8+ T Cells with Novel 68Ga-Labeled Nanobody Companion Diagnostic Agents. J. Nanobiotechnology 2021, 19, 42.
- Groeve, K.D.; Deschacht, N.; Koninck, C.D.; Caveliers, V.; Lahoutte, T.; Devoogdt, N.; Muyldermans, S.; Baetselier, P.D.; Raes, G. Nanobodies as Tools for In Vivo Imaging of Specific Immune Cell Types. J. Nucl. Med. 2010, 51, 782–789.
- Ingram, J.R.; Dougan, M.; Rashidian, M.; Knoll, M.; Keliher, E.J.; Garrett, S.; Garforth, S.; Blomberg, O.S.; Espinosa, C.; Bhan, A.; et al. PD-L1 Is an Activation-Independent Marker of Brown Adipocytes. Nat. Commun. 2017, 8, 647.
- Broos, K.; Keyaerts, M.; Lecocq, Q.; Renmans, D.; Nguyen, T.; Escors, D.; Liston, A.; Raes, G.; Breckpot, K.; Devoogdt, N. Non-Invasive Assessment of Murine PD-L1 Levels in Syngeneic Tumor Models by Nuclear Imaging with Nanobody Tracers. Oncotarget 2017, 8, 41932–41946.
- Broos, K.; Lecocq, Q.; Raes, G.; Devoogdt, N.; Keyaerts, M.; Breckpot, K. Noninvasive Imaging of the PD-1:PD-L1 Immune Checkpoint: Embracing Nuclear Medicine for the Benefit of Personalized Immunotherapy. Theranostics 2018, 8, 3559–3570.
- Bridoux, J.; Broos, K.; Lecocq, Q.; Debie, P.; Martin, C.; Ballet, S.; Raes, G.; Neyt, S.; Vanhove, C.; Breckpot, K.; et al. Anti-Human PD-L1 Nanobody for Immuno-PET Imaging: Validation of a Conjugation Strategy for Clinical Translation. Biomolecules 2020, 10, 1388.
- Broos, K.; Lecocq, Q.; Xavier, C.; Bridoux, J.; Nguyen, T.T.; Corthals, J.; Schoonooghe, S.; Lion, E.; Raes, G.; Keyaerts, M.; et al. Evaluating a Single Domain Antibody Targeting Human PD-L1 as a Nuclear Imaging and Therapeutic Agent. Cancers 2019, 11, 872.
- Liu, Q.; Jiang, L.; Li, K.; Li, H.; Lv, G.; Lin, J.; Qiu, L. Immuno-PET Imaging of 68Ga-Labeled Nanobody Nb109 for Dynamic Monitoring the PD-L1 Expression in Cancers. Cancer Immunol. Immunother. CII 2021.
- Xing, Y.; Chand, G.; Liu, C.; Cook, G.J.R.; O’Doherty, J.; Zhao, L.; Wong, N.C.L.; Meszaros, L.K.; Ting, H.H.; Zhao, J. Early Phase I Study of a 99mTc-Labeled Anti–Programmed Death Ligand-1 (PD-L1) Single-Domain Antibody in SPECT/CT Assessment of PD-L1 Expression in Non–Small Cell Lung Cancer. J. Nucl. Med. 2019, 60, 1213–1220.
- Li, D.; Zou, S.; Cheng, S.; Song, S.; Wang, P.; Zhu, X. Monitoring the Response of PD-L1 Expression to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Nonsmall-Cell Lung Cancer Xenografts by Immuno-PET Imaging. Mol. Pharm. 2019, 16, 3469–3476.
- Li, D.; Cheng, S.; Zou, S.; Zhu, D.; Zhu, T.; Wang, P.; Zhu, X. Immuno-PET Imaging of 89Zr Labeled Anti-PD-L1 Domain Antibody. Mol. Pharm. 2018, 15, 1674–1681.
- Ingram, J.R.; Blomberg, O.S.; Rashidian, M.; Ali, L.; Garforth, S.; Fedorov, E.; Fedorov, A.A.; Bonanno, J.B.; Le Gall, C.; Crowley, S.; et al. Anti-CTLA-4 Therapy Requires an Fc Domain for Efficacy. Proc. Natl. Acad. Sci. USA 2018, 115, 3912–3917.
- Lecocq, Q.; Zeven, K.; De Vlaeminck, Y.; Martens, S.; Massa, S.; Goyvaerts, C.; Raes, G.; Keyaerts, M.; Breckpot, K.; Devoogdt, N. Noninvasive Imaging of the Immune Checkpoint LAG-3 Using Nanobodies, from Development to Pre-Clinical Use. Biomolecules 2019, 9, 548.
- Lecocq, Q.; Awad, R.M.; De Vlaeminck, Y.; De Mey, W.; Ertveldt, T.; Goyvaerts, C.; Raes, G.; Thielemans, K.; Keyaerts, M.; Devoogdt, N.; et al. Nanobody Nuclear Imaging Allows Noninvasive Quantification of LAG-3 Expression by Tumor-Infiltrating Leukocytes and Predicts Response of Immune Checkpoint Blockade. J. Nucl. Med. 2021.
- Broisat, A.; Hernot, S.; Toczek, J.; De Vos, J.; Riou, L.M.; Martin, S.; Ahmadi, M.; Thielens, N.; Wernery, U.; Caveliers, V.; et al. Nanobodies Targeting Mouse/Human VCAM1 for the Nuclear Imaging of Atherosclerotic Lesions. Circ. Res. 2012, 110, 927–937.
- Sun Yoo, J.; Lee, J.; Ho Jung, J.; Seok Moon, B.; Kim, S.; Chul Lee, B.; Eun Kim, S. SPECT/CT Imaging of High-Risk Atherosclerotic Plaques Using Integrin-Binding RGD Dimer Peptides. Sci. Rep. 2015, 5, 11752.
- Chakravarty, R.; Goel, S.; Cai, W. Nanobody: The “Magic Bullet” for Molecular Imaging? Theranostics 2014, 4, 386–398.
- Jailkhani, N.; Ingram, J.R.; Rashidian, M.; Rickelt, S.; Tian, C.; Mak, H.; Jiang, Z.; Ploegh, H.L.; Hynes, R.O. Noninvasive Imaging of Tumor Progression, Metastasis, and Fibrosis Using a Nanobody Targeting the Extracellular Matrix. Proc. Natl. Acad. Sci. USA 2019, 116, 14181–14190.
- Gerdes, C.; Waal, N.; Offner, T.; Fornasiero, E.F.; Wender, N.; Verbarg, H.; Manzini, I.; Trenkwalder, C.; Mollenhauer, B.; Strohäker, T.; et al. A Nanobody-Based Fluorescent Reporter Reveals Human α-Synuclein in the Cell Cytosol. Nat. Commun. 2020, 11, 2729.
- Iljina, M.; Hong, L.; Horrocks, M.H.; Ludtmann, M.H.; Choi, M.L.; Hughes, C.D.; Ruggeri, F.S.; Guilliams, T.; Buell, A.K.; Lee, J.-E.; et al. Nanobodies Raised against Monomeric Ɑ-Synuclein Inhibit Fibril Formation and Destabilize Toxic Oligomeric Species. BMC Biol. 2017, 15, 57.
- Balhuizen, A.; Massa, S.; Mathijs, I.; Turatsinze, J.-V.; De Vos, J.; Demine, S.; Xavier, C.; Villate, O.; Millard, I.; Egrise, D.; et al. A Nanobody-Based Tracer Targeting DPP6 for Non-Invasive Imaging of Human Pancreatic Endocrine Cells. Sci. Rep. 2017, 7, 15130.
- Zheng, F.; Sparkes, A.; De Baetselier, P.; Schoonooghe, S.; Stijlemans, B.; Muyldermans, S.; Flamand, V.; Van Ginderachter, J.A.; Devoogdt, N.; Raes, G.; et al. Molecular Imaging with Kupffer Cell-Targeting Nanobodies for Diagnosis and Prognosis in Mouse Models of Liver Pathogenesis. Mol. Imaging Biol. 2017, 19, 49–58.
- Zheng, F.; Devoogdt, N.; Sparkes, A.; Morias, Y.; Abels, C.; Stijlemans, B.; Lahoutte, T.; Muyldermans, S.; De Baetselier, P.; Schoonooghe, S.; et al. Monitoring Liver Macrophages Using Nanobodies Targeting Vsig4: Concanavalin A Induced Acute Hepatitis as Paradigm. Immunobiology 2015, 220, 200–209.