Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Medicine, Research & Experimental
LncRNA PVT1 (plasmacytoma variant translocation 1) has become a staple of the lncRNA profile in patients with renal cell carcinoma (RCC).
- biomarker
- prognosis
- diagnosis
- long non-coding RNA
1. Introduction
Tumors of the kidney (renal cell carcinoma, RCC) account for 2% of all cancers [1]. Although mortality is slowly decreasing [2], RCC is considered afatal disease, with a relatively high percentage of patients having developed metastasis at the time of the diagnosis and with low chances for 5-year survival [2]. The exact identification of the RCC entity or subtype is necessary for accurate and effective treatment and proper prognosis assessment. The WHO classification system divides RCC into sixteen subtypes, the most common being three of them—clear cell carcinoma (ccRCC) with 75% abundance of all RCC cases, followed by papillary renal cell carcinoma (PRCC; 15–20%) and chromophobe renal cell carcinoma (chRCC; 5%) [3]. CcRCC is also the most aggressive type of renal cancer with the worst prognosis than the less-frequent subtypes [3,4].
Prognosis is assessed based on the TNM classification system (T—the size of the primary tumor, N—the presence of the metastases in regional lymph nodes, and M—distant metastases positivity) describing the tumor’s stage. Other prognostic information is brought in by the histologic type and level of the tumor’s differentiation [5]. From a diagnostic and prognostic point of view, it is possible to discriminate between individual RCC subtypes using immunohistochemistry or using gene expression panels [3]. With the development of targeted therapy, more attention has been turned to predictive biomarkers [6]. However, there is currently no biomarker reaching a sufficient specificity and sensitivity enabling the precise personalization of the therapy [5], although great scientific effort has been focused on searching for biomarkers in many human diseases [7], including RCC [5]. Among the potential molecules feasible as biomarkers, long non-coding RNAs have been studied excessively.
PVT1 has repeatedly emerged in many profiling studies as a prominently dysregulated lncRNA in renal cell carcinoma, even in comparison with other human tumors [10]. Therefore, we decided to summarize the current knowledge on this lncRNA and its role in the development and progression of RCC, with a particular focus on its potential feasibility as a diagnostic and prognostic biomarker.
2. Role of PVT1 in Development of Renal Cell Carcinoma
Encoded by the human PVT1 (plasmacytoma variant translocation 1) gene of nine exons, PVT1 is 1957-bp-long lncRNA located at 8q24.21 [73]. PVT1 is generally known as an oncogene involved in tumorigenesis [74]. The artificial silencing of PVT1 represses EMT and affects the proliferation, apoptosis, migration, and cell cycle by regulating many prominent tumorigenesis players, such as cyclin D1, p21, and Myelocytomatosis (MYC) [75,76,77,78]. The latter mentioned originates in a famous proto-oncogene MYC localized close to the PVT1 gene. PVT1 and MYC are known to interact with each other and are commonly coamplified [79].
Corresponding with the dysregulation in RCC tumor samples, PVT1 is overexpressed in RCC cells as well. The results show that PVT1 affects apoptosis through myeloid leukemia cell differentiation protein 1 (Mcl-1), a member of the B-cell leukemia lymphoma protein (Bcl-2) family of proteins involved in regulating cell death and comprising both pro- and antiapoptotic factors. Mcl-1 is antiapoptotic; thus, its upregulation leads to cell death signal resistance and increased survival typical for human cancer cells [80]. PVT1 knockdown in RCC cells leads to the downregulation of Mcl-1 as PVT1 enhances the Mcl-1 mRNA stability, thus keeping its levels higher (Figure 1). Downregulating either member of the PVT1/Mcl-1 axis inhibits proliferation and colony formation and promotes apoptosis [81].
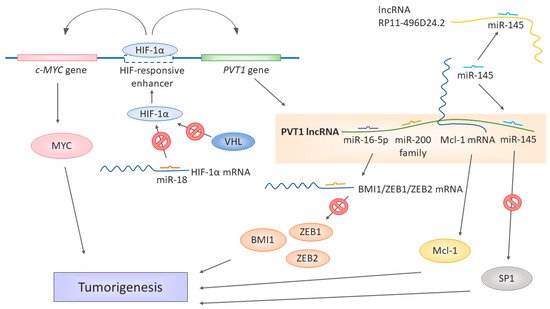
Figure 1. Different ways of lncRNA PVT1 contributing to the development of renal cell carcinoma. miR-18 binds to HIF-1α mRNA and prevents it from translation. If downregulated, the miR-18 levels are not sufficient to keep the expression of HIF-1α in check. However, while the normal oxygen levels last, protein HIF-1α is targeted by VHL for degradation. In hypoxia, HIF-1α is stabilized and cannot be targeted by VHL and can be transferred to the nucleus. The PVT1 gene is under the HIF-responsive enhancer, along with cMYC. HIF-1α binding to this enhancer activates the expression of both c-MYC and PVT1. PVT1 itself serves as a miRNA sponge binding the miR-16-5p, miR-145, and miR-200 family members, thus liberating the target mRNA from miRNA-induced degradation, for example, BMI1, ZEB1, ZEB2, and SP1. miR-145 is a target of both PVT1 and RP11-496D24.2 in the ceRNA network. Moreover, PVT1 stabilizes larger mRNA molecules such as Mcl-1, which, in turn, can be translated into functional Mcl-1 proteins. The upregulation of PVT1 is thus associated or directly responsible for the upregulation of significant factors promoting tumorigeneses, such as MYC, Mcl-1, BMI1, ZEB1, ZEB2, and SP1.
PVT1 seems to be also tightly connected to VHL signaling and hypoxia sensing [82], a significant feature of RCC tumorigenesis. On a molecular level, the most frequent cause of either sporadic or hereditary RCC is the inactivation of the von Hippel-Lindau (VHL) gene [83]. Mutations causing the loss of heterozygosity of the 3p chromosome [83,84,85] or hypermethylation of the VHL promotor [86] are common in RCC patients. Defective protein VHL enables the stabilization of the hypoxia-induced factor (HIF) protein family, which would be otherwise degraded in a proteasome. When stabilized, the HIF protein family activates and regulates the cellular response to hypoxia, affecting the genes involved in angiogenesis (for example, VEGF—vascular endothelial growth factor), cell migration, or apoptosis. VHL thus functions as a tumor suppressor [87,88].
The results of Grampp et al. [89] tie the involvement of PVT1 in MYC and VHL regulation together (Figure 1), as their team identified a unique polymorphism at 8q24.21 associated with renal cancer susceptibility. The polymorphism influences the expression of MYC and PVT1, as it is localized in the HIF-binding enhancer, affecting both the cellular MYC (c-MYC) gene and PVT1 gene. The polymorphism’s effect is restricted to renal tubular cells, and the renal cancer susceptibility depends on the genotype of the respective polymorphism site in the enhancer, affecting its accessibility to HIF proteins. pVHL defects enhanced expression of c-MYC and PVT1, hence the notorious upregulation of PVT1 in RCC. The polymorphisms in the HIF-responding regulatory elements could affect renal tumorigenesis. As the authors pointed out, the currently known SNPs associated with RCC are all linked to modulation of the VHL/HIF pathway [89,90]. Therefore, the renal cancer-promoting or protective effect of a given SNP depends on its effect on the HIF expression and HIF-mediated regulation [89].
The regulation of oxygen sensing has been expanded for the role of miR-18a [82], which regulates the expression of the HIF-1α protein. The downregulation of miR-18a leads to higher levels of the HIF-1α protein, whose expression is then unrestricted and affects the expression of PVT1 (Figure 1). The miR-18a/HIF-1α/PVT1 regulatory pathway plays a crucial role in the development and prognosis of ccRCC, as miR-18a has been identified in this work as a significant biomarker, and prognosis and RCC development [82].
The complexity of gene expression regulation by non-coding RNAs was further illustrated by Yang et al. [73] in their work showing that PVT1 also serves as a ceRNA in the context of RCC. The PVT1 levels negatively correlate with the miR-200 family and miR-20a, miR-20b, and miR-203s. PVT1 shares the miRNA-binding “seed” sequence with the miR-200-regulated mRNAs of BMI1, ZEB1, and ZEB2, all three being major players in cancer [91,92,93] (Figure 1). The subsequent in vitro experiments showed that PVT1 upregulates the levels of BMI1, ZEB1, and ZEB2, while the overexpression of the miR-200s downregulated them. The authors further identified a new splicing variant of PVT1, whose levels were even higher in the tumor tissue and tumor cell lines than the levels of the full-length original splicing variant and had an even higher effect on the cell proliferation. The spliced region, exon 4, does not contain binding sites for the miR-200 family. Therefore, even this variant can act as ceRNA [73]. Ren et al. [77] identified the functional connection of PVT1 with miR-16-5p, as the silencing of this miRNA has led to the reversion of the regulatory effect on RCC cells induced by the downregulation of PVT1. An in-silico target prediction showed that miR-16-5p is a target of PVT1. Thus, its levels decrease as a result of binding to PVT1 (Figure 1). Moreover, the strong negative regulatory connection of PVT1 and miR-16-5p has been observed in colorectal cancer [94].
A similar ceRNA mechanism has been observed in PRCC by Huang et al. [95]. In their work, they found the interactions of miR-145 miR-211, miR-216a, mIR-133a, and miR-133b with PVT1. These interactions overlapped with lncRNA RP11-496D24.2, which also contained binding sites for some of these miRNAs (mir-145, mir-211, and mir-216a). The authors suggested a competition mechanism between PVT1 and RP11-496D24.2 for these common miRNAs, thus being critical regulators of pathogenesis in PRCC. However, the downstream targets profiting from RP11-496D24.2-mediated miRNA binding are not yet known. In PVT1, though, SP1 has been identified as a potential target of PVT1, miR-145, miR-133a, and miR-133b. SP1 is a versatile transcription factor [96] involved in many cellular processes as an activator or repressor, depending on the context. Suggested lncRNA-miRNA-mRNA ceRNA pathway regulating of such a potent regulator of gene expressions could be a significant driving force in the pathogenesis of PRCC, especially with the upregulation of PVT1, which was shown by Huang et al. [95] in PRCC patients.
The complex system of PVT1 tumorigenesis regulation via the regulation of miRNA was exceptionally summarized in the work of Wang et al. [97]. PVT1 functions as a miRNA “sponge”, inhibiting their activity by lowering the levels of more than 20 miRNAs. Should those miRNAs be involved in the regulation of other oncogenic mRNAs, the upregulation of PVT1 would lead to their decrease and, thus, the promotion of tumorigenesis. Moreover, PVT1 seems to be a precursor to other miRNAs with either oncogenic or tumor-suppressive effects [97].
This entry is adapted from the peer-reviewed paper 10.3390/biom11050664
This entry is offline, you can click here to edit this entry!