As widely acknowledged, 40–50% of all melanoma patients harbour an activating BRAF mutation (mostly BRAF V600E). The identification of the RAS–RAF–MEK–ERK (MAP kinase) signalling pathway and its targeting has represented a valuable milestone for the advanced and, more recently, for the completely resected stage III and IV melanoma therapy management. However, despite progress in BRAF-mutant melanoma treatment, the two different approaches approved so far for metastatic disease, immunotherapy and BRAF+MEK inhibitors, allow a 5-year survival of no more than 60%, and most patients relapse during treatment due to acquired mechanisms of resistance. Deep insight into BRAF gene biology is fundamental to describe the acquired resistance mechanisms (primary and secondary) and to understand the molecular pathways that are now being investigated in preclinical and clinical studies with the aim of improving outcomes in BRAF-mutant patients.
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
The identification of signalling pathways and tumour immune microenvironment interactions has revolutionized the treatment of melanoma [
1]. To date, two largely mutually exclusive groups of cutaneous melanomas can be categorised: those harbouring an activating BRAF mutation (mostly BRAF V600E), which represent 40–50% of all melanoma patients, and those harbouring other mutations than BRAF [
2]. Indeed, as widely acknowledged, the identification of the RAS–RAF–MEK–ERK (MAP kinase) signalling pathway and its targeting has represented a valuable milestone for the advanced and, more recently, for the completely resected stage III and IV melanoma therapy approaches [
3]. In patients with unresectable or metastatic disease, the therapeutic available strategies of targeted therapy (TT) with BRAF inhibitor (BRAFi) and MEK inhibitor (MEKi) and immune check-point inhibitors (ICIs) have led to a significant improvement in overall survival (OS) and progression-free survival (PFS). However, despite the recognized progress, the two different approaches approved so far for metastatic disease allow a 5-year survival of no more than 60%, even with important differences according to first-line drug(s) used and prognostic factors; moreover, most patients relapse during treatment due to acquired mechanisms of resistance [
4,
5]. Deep insight into the BRAF gene biology is fundamental to further describe the resistance mechanisms (both primary and secondary) and to understand the molecular pathways that are now being investigated in preclinical and clinical studies with the aim to improve outcomes in BRAF-mutant patients.
2. BRAF Gene Biology
The BRAF gene is located on chromosome 7 (7q34) and encodes the BRAF protein, a 94 kDa intracellular enzyme of 766 amino acids involved in the mitogen activated protein kinase (MAPK) pathway [
6]. The MAPK pathway consists of a chain of intracellular proteins which regulates physiological cell functions including growth, differentiation, proliferation and apoptosis [
7]. BRAF, as well as ARAF and CRAF, is a MAPK kinase kinase (MAPKKK), and it is typically activated by GTPases proteins—namely, RAS proteins—downstream from cell surface receptors, such as EGFR (Epidermal Growth Factor Receptor) or KIT, even if several more kinds of stimuli could lead to its activation [
8].
BRAF protein consists of three conserved regions (CRs): CR1, which is composed of a RAS-binding domain (RBD) followed by a cysteine-rich domain; CR2 is a serine/threonine-rich domain containing a 14-3-3 binding site; and CR3 is the catalytic serine/threonine protein kinase domain [
9]. To be effective, BRAF must dimerize in order to form complexes with MEK, adjuvated by 14-3-3 proteins; the latter ones are involved in both active and inactive phases of BRAF signalling [
10].
Once activated, BRAF phosphorylates mitogen-activated protein kinase/extracellular signal-regulated kinase ERK kinase (MEK) which, in turn, phosphorylates the extracellular signal-related kinases 1 and 2 (ERK1/2) [
11]. ERK proteins are the last effectors of the pathway: once phosphorylated, they dimerize and translocate into cell nucleus, thus activating, through phosphorylation, many transcription factors, such as c-Jun and c-Myc [
12]. The final goals of this signalling pathway in physiological conditions are the control of cell cycle progression and the regulation of apoptosis [
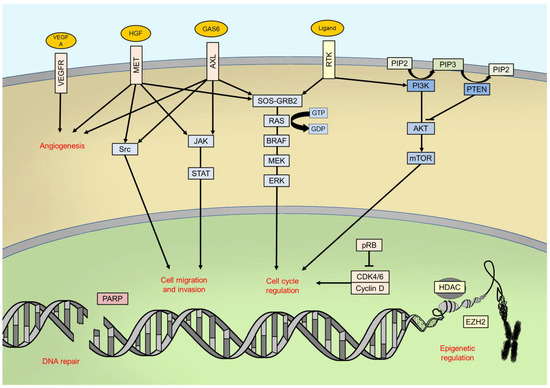
11] ().
Figure 1. Main intracellular pathways and processes involved in melanomagenesis. RTK: receptor tyrosine kinase; PIP2: phosphatidylinositol 4,5-bisphosphate; PIP3: phosphatidylinositol (3,4,5)-trisphosphate.
3. BRAF Mutations in Melanoma: Epidemiology and Clinic-Pathological Correlations
Since the early 2000s, BRAF has been identified as a commonly mutated gene in human tumours [
2]. Mutations in the BRAF gene could cause an impaired protein function, depending on localization and type [
13].
Concerning cutaneous melanoma, the most frequent (65%) and relevant alterations in BRAF gene sequence are those affecting codon V600 (formerly named V599) in the exon 15 [
2,
14]. BRAF V600 mutations have been detected in nearly 50% of all cutaneous melanoma patients [
15]. Non-V600 mutations, which are less frequent than V600 ones, have been found in 11% of all cutaneous melanoma patients [
14].
BRAF mutations in cutaneous melanoma are most common on the trunk (affecting less frequently the head and neck), on skin without marked solar elastosis and in younger age, thus suggesting a physiopathology role for intermittent UV exposition in early life rather than chronic sun damage [
16]. A recent study using sequencing data showed a model of the propagation and selection of clones with different categories of BRAF mutations to establish their evolutionary trajectories. The phylogenetic trees of cutaneous melanoma samples with amplification of BRAF express a major dominant clone, with only rare intermediates that are persistent from the previous selective sweeps, consistent with a linear evolutionary process. However, it is still not clear whether melanoma with amplification of BRAF experiences iterative selective sweeps and, if so, what the underlying molecular basis of this process might be [
17].
Actually, clinicopathological characteristics and frequency are different for each kind of BRAF mutation, so we should consider them as different entities.
3.1. BRAF V600 Mutations
BRAF V600E is, globally, the most frequent mutation observed in cutaneous melanoma patients, accounting for 70–88% of all known V600 BRAF mutations [
16]. It consists of an amino acid change from valine (V) to glutamic acid (E), resulting in a 480-fold increase in kinase activity (catalytically active conformation) compared with native protein [
13]. BRAF V600K is the second most common mutation (10–20% of all V600 BRAF mutations) in cutaneous melanoma and, as V600E mutation, it consists of an amino acid change, with a valine (V) replaced by a lysine (K) [
18]. Other rarer codon V600 BRAF mutations, approximately 10% of all V600 mutations, are V600R (<5%), V600D (<5%), V600E2 (<1%), V600M (<1%) and V600G (<1%) [
17,
19,
20]. Cutaneous melanomas harbouring BRAF V600E and V600K mutations, even if similar from a molecular point of view, have distinct clinicopathological features (). In fact, BRAF V600K-mutant cutaneous melanomas are considered more aggressive than V600E ones, since they have shown less tumour regression and shorter progression-free survival during treatment with combined BRAF and MEK inhibitors, together with a shorter disease-free interval from diagnosis of primary melanoma to the occurrence of first distant metastasis [
21,
22]. Analysis of BRAF V600K-mutant cutaneous melanoma samples from the Cancer Genome Atlas highlighted, with respect to V600E, an upregulation of energy metabolism, emphasizing their clinical aggressiveness [
23]. On the other hand, an older age at diagnosis, a higher degree of cumulative sun-induced damage and a higher mutational burden have been described in V600K-mutant cutaneous melanoma with respect to V600E melanomas, explaining good response to immunotherapy [
21,
22].
Table 1. Different clinicopathological characteristics between BRAF V600E- and V600K-mutant melanoma patients.
Concerning prognosis, BRAF V600 mutations are statistically significantly associated with reduced OS in cutaneous melanoma patients [
24]. In fact, it must be stressed that there is a higher trend of BRAF V600-mutant melanoma, with respect to BRAF wild-type ones, to involve the brain and liver as a first site of metastasis, thus affecting negatively the prognosis of these patients [
25]. BRAF V600 mutations also have an important predictive role, since metastatic cutaneous melanoma patients harbouring them do respond to specific inhibitors, as further described. Rare V600 BRAF mutations, such as V600R, V600D and V600M, have been associated with good response to BRAF inhibitors and acceptable OS compared to V600E/K-mutant melanoma patients [
26].
3.2. BRAF Non-V600 Mutations
BRAF non-V600 mutations, as stated before, are less frequent than V600 ones, and their prognostic and predictive role is, to date, still difficult to elucidate.
L597, K601 and G469 mutations, also known as class II BRAF mutations, determine an increased kinase catalytic activity, different from monomeric V600-mutant protein, through constitutive dimerization. Interestingly, they are located in different regions of the gene: L597 and K601 in the activation segment, whilst G469 in the glycine rich region of BRAF [
14]. Even if these mutations do not confer sensitivity to BRAF inhibitors, they activate downstream target proteins, thus explaining sensitivity to MEK inhibitors [
27].
Codon D594 and G596 mutations, also known as class III BRAF mutations, have been described as kinase-impairing alterations [
28,
29]. In fact, different from V600E mutations, which cause hyperactivation of downstream kinase pathways, kinase-impairing mutations cause a reduction in BRAF catalytic activity; these proteins are RAS dependent and have low or absent kinase activity [
14,
30]. Codon D594 and G596 mutations are rare, < 4% of all melanoma patients, but have been associated with a good prognosis and a more prolonged OS than V600-mutant patients [
30]. Codon N581 mutations, very rare, are also kinase-impairing mutations [
29].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22073474