In the body, CD38, a multifunctional glycoprotein enzyme present on the surface of immune cells, converts the pyridine nucleotides NAD and NAAD into ADPR, but NADP and NAADP into ADPR-2-phosphate (ADPRP) [
38]. These nucleotides and others, the metabolism of which is intertwined with that of ADPR, were tested to address the potential effects on TRPM2. NAD was identified as a channel agonist in early studies [
5,
28], while no binding to the NUDT9-H domain was later reported [
24]. NAAD [
31], NAADP [
31,
39,
40] and cADPR [
39,
40] were reported to act as activators, AMP as an inhibitor [
24,
26], while NADH and NADP were found to be ineffective [
5]. Besides these reported effects on channel activity, cADPR [
26,
39,
40] and NAADP [
39,
40] were also shown to augment the effects of ADPR in a positively cooperative manner. 8-Br-cADPR, an agent shown to reduce ischæmic acute kidney injury [
39], was suggested to act as a partial TRPM2 agonist and a competitive inhibitor of activation by cADPR [
26]. Most of the above studies examined the effects of nucleotides in whole-cell systems. In a study in inside-out patches, the effects of cADPR on TRPM2 channel activity were shown to be attributable to ADPR contamination [
31]. Decontamination of commercial cADPR stocks with a nucleotide pyrophosphatase which selectively cleaves ADPR but not cADPR eliminated channel stimulation by the compound in excised patches. Furthermore, no cooperative effects of cADPR with ADPR were observed. Recently, Yu et al reported that synthetic pure cADPR directly binds to the human NUDT9-H domain [
41]. SPR titration indicated an apparent affinity (K
d) of ~12 μM; however, much higher concentrations of cADPR (EC
50 ~250 μM) were required to stimulate macroscopic TRPM2 currents. Considering its submicromolar cellular concentration [
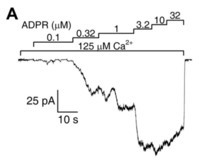
42], cADPR does not seem to act as a physiological activator of TRPM2. TRPM2 channel activation by various pyridine dinucleotides is explained by their spontaneous degradation, which also produces ADPR(P). Cleavage of contaminating ADPR in NAD and NAAD stocks, or of contaminating ADPRP in NAADP stocks, by the purified NUDT9 ADPRase abolished the stimulatory effects of the dinucleotides, ruling them out as direct effectors of TRPM2 [
43]. Moreover, these experiments also demonstrated that none of the spontaneous and/or enzymatic cleavage products—nicotinamide, nicotinic acid, AMP(P) or ribose-5-phosphate—are TRPM2 channel activators. An apparent stimulatory effect of NAADP prior to decontamination pinpointed its spontaneous degradation product ADPRP as an agonist. Indeed, in inside-out patches, pure ADPRP opened TRPM2 channels, albeit its apparent affinity was lower (EC
50~13 μM) and maximal stimulation smaller (~80%) when compared to ADPR. The lower open probability supported by ADPRP was explained by an approximately three-times faster macroscopic closing rate, reflecting a shortened open burst [
43]. A similar efficacy but even lower affinity (EC
50~110 μM) for ADPRP was reported in a whole-cell study [
44] that also showed 2’-deoxy-ADPR (dADPR) to act as a superagonist on hsTRPM2 channels. In the presence of dADPR, whole-cell TRPM2 currents were 37% larger, and following patch excision channels inactivated slower, but the EC
50 of dADPR was similar to that of ADPR. Since upon oxidative stress-induced DNA damage, dADPR may accumulate due to CD38 activity, this nucleotide is a likely regulator of TRPM2 function under pathophysiological conditions.