Lung cancer is a well-known malignant tumor of the respiratory tract, which has caused a significant level of damage to human health in the 21st century. Micro-RNAs (miRNAs) are tiny, non-coding RNA stem-loop structures with a length of roughly 20–25 nucleotides that function as powerful modulators of mRNA and protein products of a gene. miRNAs may modulate many biological processes involving growth, differentiation, proliferation, and cell death and play a key role in the pathogenesis of various types of malignancies. Several accumulating pieces of evidence have proven that miRNA, especially miR-146a, are crucial modulators of innate immune response sequences. Changes in miR-146a expression levels have biomarker importance and possess a high potential as a therapeutic target in lung cancer. It retards epithelial-mesenchymal transition and promotes the therapeutic action of anticancer agents in lung cancer. Studies have also suggested that miR-146a affects gene expression through different signaling pathways viz.
- miR-146a
- lung cancer
- biomarker
- clinical pathology
1. Lung Cancer and miR-146a
Lung cancer is one of the most common malignancies worldwide (11.6%) and accounts for nearly 18.5% of cancer-related mortality. About 2,093,876 new cases of lung cancer are diagnosed per year, with 1,761,007 deaths [1]. Small cell lung cancer (SCLC) is characterized as a neuroendocrine carcinoma because the cancer cells have features of nerve cells and endocrine (hormone-secreting) cells and is less prevalent (16.8%) but more lethal and heterogeneous than non-small cell lung cancer (NSCLC) among the population (80.4%) [2]. Lung carcinogenesis is a multifactorial and multistep process that causes sequential accumulation of molecular and genetic defects, especially because of tobacco use. It initiates with the loss of 9p and 3p chromosomes and ends with cyclin D1 and E overexpression [3][4][5]. Takamizawa et al. first observed non-coding RNA dysregulation in lung cancer when he noticed a significantly reduced level of let-7 associated with the post-operative survival of lung cancer patients [6]. Lin-4 became the first miRNA discovered when R. C. Lee was studying postembryonic developmental mechanisms in C. elegans [7]. These are a group of small non-coding RNAs with an excellent function in post-transcriptional regulation of gene expression. The two ways by which miRNA monitors gene expression are by arresting mRNA translation or by mRNA degradation [7]. Since malignancy accounts for highly heterogenous diseases, analysis of miRNA expression can discriminate among subtypes of cancer [8][9][10][11] and has the potential to detect the unknown origin site of primary cancer [12]. miRNAs can serve as good candidates to predict the clinical outcome or clinical progression of cancer, including its survival rate, severity [13], and development of chemoresistance [14][15][16].
Dr. David Baltimore found that the gene for miR-146a was located on chromosome 5 [17][18]. miR-146a was found to facilitate programmed cell death and discourage cell proliferation and cell migration in NSCLC cell lines that were the key hallmarks of cancer [19]. miR-146a also showed downexpression, which was proven in several NSCLC cell lines [20] and significantly associated with advanced-stage lung cancer that lowered progression-free survival [19].
2. miR-146a Expression and Regulation
2.1. Biogenesis
The regulatory role of miRNAs was not well known until 2001, after which thousands of miRNAs in diverse kinds of species were discovered [6][21]. RNA polymerase II (Pol II) is the main enzyme that catalyzes most of the miRNA gene transcription from the dedicated miRNA gene loci in the nucleus, and around 30% are generated from protein-coding gene introns. The resultant primary miRNAs (pri-miRNAs) are further processed by capping, splicing, and polyadenylation [7]. A single miRNA or a group of miRNAs are produced by these pri-miRNAs from a common primary transcript. The long pre-miRNAs are cleaved by microprocessors, which are a complex of dsRNase III enzyme (DROSHA) and a cofactor called double-stranded RNA (dsRNA) binding protein vital region 8 (DGCR8) [22][23]. The two dsRNA strands of pri-mi-RNAs are cleaved toward the base of secondary stem-loop structures by the two RNA-III domains found in DROSHA, which release hairpin-shaped precursor miRNAs (pre-miRNAs; ~60–70 nucleotide) [23][24][25]. The single-stranded RNA (ssRNA) stem junction and the distance from the terminal loop region are then recognized by the microprocessor. In particular, the microprocessor slices the dsRNA at ~10–12 bp from the juncture with flanking ssRNA, thereby creating hairpin-shaped pre-miRNAs with an overhang of either two nucleotides (group I miRNAs) or one nucleotide (group II miRNAs) at the 3′ end [26][27][28][29]. Since the essential elements, DROSHA and DGCR8, are necessary for the biogenesis of virtually all cell’s miRNAs, in vitro microprocessor activity contained in recombinant DROSHA and DGCR8 proteins can be reconstituted [30]. Many auxiliary elements are believed to play a role in the production of cells’ pri-miRNA. Exportin 5 (XPO5) then transports the pre-miRNAs to the cytoplasm from the nucleus. This is further processed by DICER1, an RNase III enzyme measuring the 5′ and 3′ pre-miRNA ends. DICER1 binds to the end of the pre-miRNA and positions its two catalytic RNase III domains such that the mature ~22-nucleotide miRNA duplex with 2-nucleotide 3′ overhangs is formed by asymmetrical cleavage of the dsRNA stem close to the terminal loop sequence. Transactivation-responsive RNA-binding protein (TRBP; also known as TARBP2) that binds to dsRNA is then associated with DICER1 [31][32]. While DICER1 does not involve pre-miRNA processing, TRBP increases the accuracy of DICER1-mediated cleavage of pre-miRNAs in a structure-dependent manner and modifies the selection of miRNA guide strands by leading to iso-miRNA formations that are one nucleotide longer than the normal miRNAs [31]. The other function of TRBP is to bridge DICER1 and argonaute proteins (AGO1, AGO2, AGO3, or AGO4) physically to participate in the miRNA-induced silencing complex (miRISC) assembly [32]. Argonaute protein binds to one strand of the mature miRNA, which is the guide strand, and leads the complex to target complementary mRNAs with GW182 protein family members for post-transcriptional gene silencing. This happens in processing bodies (P-bodies) where the cytoplasmic foci are induced by mRNA silencing and decay but are not usually necessary for the silencing of miRNA-mediated genes [33][34] (Figure 1).
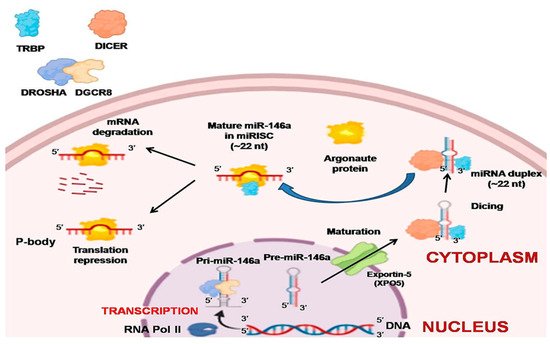
Figure 1. Biogenesis of miR-146a. Pri-miR-146a is formed by RNA polymerase II, which is further processed by capping, splicing, and polyadenylation. The long pre-miR-146a is cleaved by the microprocessor to release a hairpin-shaped pre-miR-146a. This leaves the nucleus through exportin 5 (XPO5) into the cytoplasm. In the cytoplasm, DICER with transactivation-responsive RNA-binding protein (TRBP) further acts on the pre-miR-146a to generate miRNA duplex. Argonaute protein selectively binds to one of the strands of miRNA, and a miRNA-induced silencing complex (miRISC) assembly is formed. The strand to which argonaute binds serves as a guide strand and leads the complex to complementary target mRNAs with GW182 protein family members for post-transcriptional gene silencing forming processing bodies (P-bodies). The other fate of miRNA is degradation if it is not needed.
2.2. Transcriptional and Post-Transcriptional Regulation
Throughout vertebrate species, miR-146a (mature) is greatly conserved [35]. As discussed above, the pri-miRNA-146a is produced as an independent unit that is regulated by a unique regulatory sequence. As shown by the experimentation of Taganovet et al. (2006), this regulatory sequence is located 16 kb upstream from the miR-146a gene. This regulatory sequence behaves as an interacting sequence for specific transcription activators or repressor factors as one site for C/EBPβ, IRF3/7, and two for NF-kB. Any kind of inflammatory signaling pathway that is mediated via the NF-kB factor strongly upregulates miR-146a expression such as lipopolysaccharide (endotoxin) from Gram-negative bacteria, TNF-α, and IL-1β signaling pathways [18]. In melanoma cells, apart from NF-kB, researchers also found that the c-MYC site increases miR-146a expression [36][37].
Molecular mechanisms, such as CpG methylation in the promoter or histone deacetylations, lead to downregulation of miR-146a in cancers [38]. For instance, CpG methylation on the miR-146a regulatory sequence causes its downregulation in NSCLC cell lines [20], which is also observed in hepatocellular carcinoma (HCC) [39] and prostate cancer cells [40]. H3 histone deacetylation at NF-kB sites leads to downregulation of miR-146a in macrophages and experiences significant improvement in miR-146a expression when treated with HDAC inhibitor. It is interesting to note that long non-coding RNA (lncRNA) directly interacts with miR-146a and significantly interferes with its gene-silencing function since lncRNA possesses multiple miRNA binding sites that trap miRNA and prevent its biological function [41]. For instance, lncRNA NIFK-AS1 in tumor-associated macrophages (TAMs) traps miR-146a and promotes Notch1 signaling in endometrial malignancies [42]. According to a similar study, in which lncRNAs like SNHG16, whose upregulation causes miR-146a downregulation, shows lncRNA also facilitates the progression of NSCLC [43]. Kumaraswamy et al. found the restoration of BRCA1 expression in breast cancer significantly decreases EGFR levels as well as restores miR-146a expression. It is confirmed that BRCA1 directly binds the miR-146a promoter and increases its expression, which in turn represses EGFR levels [44]. By studying malignant breast cells (MCF7) and normal epithelial breast cells (MCF10A), Lie et al. observed forkhead box protein P3 (FOXP3) binding sites in the miR-146a promoter that regulates its expression at the transcriptional level [45]. One study based upon chromatin immunoprecipitation (ChIP) suggests that the signal transducer and activator of transcription 3 (STAT3) directly binds to the promoter region of miR-146a, but no relevant binding site positions are described by the authors [46]. In summary, the expression of miR-146a is regulated by various regulatory proteins, especially transcription factors, as well as by epigenetic modifiers and lncRNA in various cells. These experiments have demonstrated that, in different types of cells, various regulatory proteins and transcription factors are responsible for miR-146a regulation. However, modifications and alterations of these mechanisms in cancer cells contribute to the misexpression of miR-146a. When Larner-Svensson et al. used pharmacological inhibitors on human airway smooth muscle cells, he observed that the expression of IL-1β-induced miR-146a was controlled at the transcriptional level by activating IKK2 (i.e., by NF-κB activity) but the pathway that produced mature miR-146a from the primary transcript was regulated by the mechanism-dependent modulation of MEK-1/2 and JNK-1/2 [47] (Figure 2).
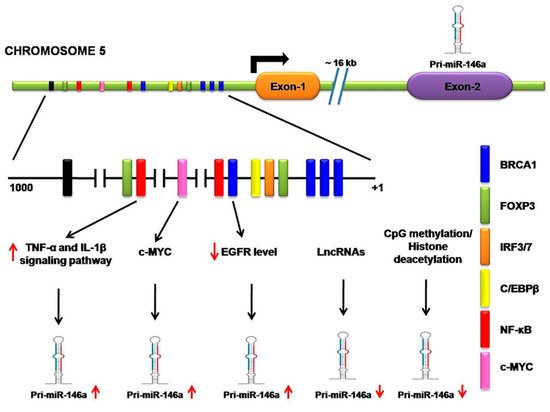
Figure 2. Transcriptional and posttranscriptional regulation of miR-146a. A regulatory sequence is located 16 kb upstream from the miR-146a gene. It contains a site for several transcription activator or repressor factors, such as breast cancer 1(BRAC1), forkhead box protein P3 (FOXP3), interferon regulatory transcription factor (IFR3/7), CCAAT enhancer-binding proteins β (C/EBPβ), nuclear factor kappa-B (NF-κB), and c-MYC. NF-κB regulatory inflammatory signaling pathways strongly upregulate miR-146a. c-MYC also increases miR-146a expression. BRCA1 directly binds the miR-146a promoter, increasing its expression, which in turn represses EGFR levels. Long non-coding RNA (lncRNA) directly interacts with miR-146a and significantly interferes with its gene-silencing function. CpG methylation on the miR-146a regulatory sequence causes its downregulation.
3. Role of miR-146a in Lung Cancer
As observed by Bertoli and coworkers, different groups of miRNAs play an important role in different hallmarks of breast cancer [48]. It has been suggested that miR-146a may play an important role in several hallmarks of lung cancer, such as evading growth barricades, cell proliferation, resisting cell death, promoting angiogenesis, inflammation, tumor immune tolerance, energy metabolism imbalance (Warburg effect), metastasis, and genome fragility, that were first explained by Hanahan and Weinberg [49]. Since miRNA dysregulation has been found in almost every type of tumor, their involvement in cancer hallmarks is likely. Apart from lung cancer hallmarks, miR-146a involvement in cancer stem cells (CSCs) has also been observed. There is a similarity between EMT and CSC formation since both have several common signaling mediators like Notch, Wnt, and Hedgehog proteins. Many studies have found stem cell-like properties, thus sharing key signaling pathways and drug resistance phenotypes with CSCs [50][51].
3.1. miR-146a as an Antiproliferative and Proapoptotic Agent
Most of the research associated with miR-146a indicates its tumor-suppressive role in malignancies. In NSCLC cell lines and human lung tissue samples, the expression of this miRNA is strongly downregulated and it also displays antiproliferative and antiapoptotic properties in lung cancer cell lines [52][53]. The gene for EGFR found to be commonly mutated in lung adenocarcinoma patients, especially in nonsmoking women with Asian ethnicity (50%) [54][55], is directly targeted by miR-146a [19]. Qi et al. (2019) recently found that miR-146a-5p directly targets EGFR mRNA while studying the effect of cryptotanshinone derived from Salvia miltiorrhiza on NSCLC. It was also observed that cryptotanshinone prevents cell cycle progression by upregulating miR-146a/b levels [56]. The tumor collagenase stimulatory factor, also known as the extracellular matrix metalloproteinase inducer (EMMPRIN), is expressed on the outer surface of human tumor cells. This glycoprotein interacts with the fibroblasts and triggers many matrix metalloproteinase expressions within the fibroblasts [57]. Huang WT et al. proved through various study methods like TargetScan, luciferase enzyme assay, cancer genome atlas, and immunohistochemistry that miR-146a directly targeted the tumor collagenase stimulatory factor (TCSF) to modulate the mechanisms of cell viability and apoptosis in NSCLC [58]. Macrophage migration inhibitory factor (MIF) was considered an important cytokine for the regulation of innate immunity. Thorsten Hagemann et al. knocked down either EMMPRIN or MIF and found decreased invasiveness and matrix metalloproteinase activity in the supernatant of tumor cell culture [59]. MIF was also found to be the promoter of the Warburg effect in NSCLC by activating NF-kB/HIF-1α inflammatory signaling [60][61]. The macrophage migratory inhibition factor (MIF) gene upregulated in NSCLC is the reverse target of miR-146a, as proven by luciferase assay mimic studies in A549 cells, and promotes apoptosis and discourages proliferation [62]. Another similar study found cell cycle progression was also slowed down by miR-146a via specifically downregulating the expression of CCND1/2 (genes of cyclins D1/2), both at post-transcriptional and protein stages that arrested the G0/G1 phase of cell cycle progression [63] and targeted cyclin J that promoted NSCLC chemosensitivity to cisplatin [64]. It was found that miR-146a overexpression maintained epithelial phenotypes and discouraged epithelial to mesenchymal transition in lung cancer cell lines by suppressing insulin receptor substrate-2 (IRS2) transcription and translation [65]. It has also been found that IRS2 enhances the Wnt/β-catenin pathway, thus increasing N-cadherin but decreasing E-cadherin [66]. This was further confirmed by a study of a xenograft mouse model where miR-146a overexpressing cells decreased the tumor sizes. Downexpression of miR-146a was recently found to be caused by some epigenetic cellular events like promoter hypermethylation [20] and post-transcriptional silencing by competitive endogenous RNA (ceRNA) in NSCLC. It was also recently found that SNHG16, a competitive endogenous RNA (ceRNA), was associated with poor prognosis and upregulation in NSCLC [43]. However, the above observations were contradicted by Tan et al., who observed miR-146a overexpression both in vivo and in vitro experimental systems such as malignant lung tissue and NSCLC cell lines. He experimentally justified miR-146a as an oncomer since CHOP (DNA damage-inducible transcript 3) was targeted by miR-146a, whose downexpression has been linked to poor prognosis in lung cancer [67]. The Merlin/NF2 tumor suppressor protein level was proven to be negatively regulated by miR-146a by directly interacting with its mRNA and triggering cell proliferation, invasion, and cell migration by miR-146a in vivo cell transfection studies in A549 lung epithelial cells [68]. However, a large body of evidence proved the tumor-suppressive role of miR-146a in NSCLC.
3.2. miR-146a as an Anti-Inflammatory Agent
The strong association between cancer and chronic inflammation was first discovered by Virchow in 1863 [69]. When there is an inflammatory insult such as a malignant lesion, leukocyte cells that produce a diverse variety of cytokines, chemokines, and other inflammatory molecules, will be attracted. It is a well-recognized fact that inflammation promotes cell proliferation, immune protection of malignant cells, angiogenesis, and cell metastasis [70]. There is also a strong link between COX2 and initiation of carcinogenesis [71]. One of the well-recognized miRNAs, miR-146a, is the key inflammatory signaling homeostasis and innate immunity molecules through target genes IRAK1 and TRAF6 that are downstream mediators of the IL-1α and TNFα signaling pathways. It seems that miR-146a prevents cytokine overproduction following bacterial endotoxin challenges or any inflammatory insults, and continued expression is associated with immune tolerance that fine-tunes the inflammatory system in order to stop inflammatory response overstimulation [72]. The overexpression of miR-146a in lung cells causes negative regulation of the metabolism of arachidonic acid via directly targeting the two key nodes of the inflammatory pathway: cyclooxygenase-2 (COX-2) and 5-lipoxygenase-activating protein (FLAP) [20]. This helps to prevent the overproduction of miR-146a prostaglandins E2 (PGE2) and leukotriene B4 (LTB4) that are potent inflammatory agents and promote promalignant microenvironments. Thus, it is apparent that underexpression of miR-146a leads to upregulation of COX-2 and FLAP proteins, which causes overproduction of inflammatory effector molecules such as PGE2 and LTB4 in NSCLC cell lines. This inflammatory microenvironment serves as a suitable site for the initiation of tumorigenesis. The signaling of IL-1β induces the secretions of inflammatory cytokines, such as IL-8 and RANTES. Taganov et al. found that IL-1β signaling increased miR-146a expression 24 times via the NF-κβ transcription factor [18], but when human lung alveolar epithelial cells were transfected with miR-146a mimics, significant IL-1β-induced IL-8 and RANTES inhibition was observed. While they neither targeted their release nor transcription, it was suggested that they destabilized their mechanism of translation [73]. Lambert KA et al. observed the activation of miR-146a expression as a protective negative feedback mechanism induced by inflammatory conditions to decrease inflammation. Codelivery of miR-146a and anti-inflammatory agents such as glucocorticoids enhanced their therapeutic action and thus could be proven to be a unique therapeutic strategy to reinforce the efficacy of these medications [74]. It was found by Limin that particulate matter-induced inflammation caused activation of miR-146a expression along with IL-6 and IL-8 in BEAS-2B cells. In turn, miR-146a blocked the transport of p65 towards the nucleus through inhibition of IRAK1/TRAF6 and prevented the release of IL-6 and IL-4 [75]. Thus, miR-146a also serves as a negative feedback mechanism to protect us from exogenous inflammatory agents that may trigger the development of lung cancer. Senescence is a cellular program that irreversibly prevents impaired cell proliferation and activates the release of IL-6 and IL-8, which are part of a larger senescence. Scott et al. reported the upregulation of miR-146a/146b to be allied to the senescence-associated secretory phenotype (SASP) as compared to quiescent fibroblast cells [75]. It was also observed that IL-1α signaling stimulated miR-146a/146b expression, which in turn negatively regulated IL-1α induced cytokine secretion. Therefore, it seems that miR-146a/146b expression upregulation responds to the situation of elevated inflammatory cytokine levels within a cell as a protective negative feedback, such as senescence-associated SASP activity [75].
3.3. miR-146a as a Metastatic Modulator
Metastasis is the migratory process of primary tumor cells to distant regions that becomes a dominant cause of death by cancer, such as in non-small cell lung cancer (NSCLC) [76][77]. Metastasis starts with a breach of physical barricades, such as the basement membrane, invading neighboring tissues and lymph nodes, and circulating into the blood and lymph. It traverses the blood vessel walls to arrive at its next destination and, finally, results in the proliferation of a secondary tumor [78][79]. A number of emerging studies found that microRNAs played a pivotal role in epithelial–mesenchymal transition (EMT), an important starting step in the development of metastasis. For instance, miR-155 enhanced the process of EMT by directly downexpressing RhoA GTPase, an important player in cell polarity, development, and maintenance of tight junctions [80]. However, after analysis of many studies, it seems that miR-146a plays a dual role (inhibitory and stimulatory) in metastasis [81]. It discourages metastasis and cell invasion by weakening NF-kB signaling and its associated targets in liver cancer [39], breast cancer [82], and colorectal cancer [83]; while its increased level in oral squamous cell carcinoma enhances stemness potential by protecting β-catenin from proteasomal degradation and downexpressing E-cadherin and CD24 transcriptions [84]. Chemoresistance and metastasis are two interwoven processes, and chemoresistance may be the trigger for metastasis [85]. Many studies found that miR-146a promoted cisplatin sensitivity or reduced cisplatin resistance and thus regressed metastasis in lung cancer via certain molecular targets already discussed in the manuscript. However, a recent study found that miR-146a significantly overexpressed, along with a reduced expression of tumor suppressor Merlin protein, and experimentally proved its direct target in lung adenocarcinoma [86] (Figure 3).
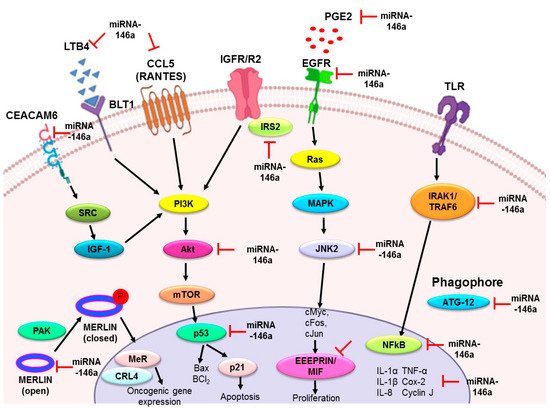
Figure 3. Summary of the role of miR-146a in lung cancer. The upregulation of miR-146a in lung cancer has a preventive role by acting as an antiproliferative and proapoptotic agent, anti-inflammatory agent, and metastatic modulator. Various downstream targets mentioned in the figure have been identified in different in vitro and in vivo models, justifying these properties of miR-146a. EGFR: epidermal growth factor receptor; EMMPRIN: extracellular matrix metalloproteinase inducer; MIF: macrophage migration inhibitory factor; IRS2: insulin receptor substrate 2; Merlin/NF2: moesin-ezrin-radixin-like protein/neurofibromatosis type 2; IRAK1: interleukin-1 receptor-associated kinase 1; TRAF6: tumor necrosis factor receptor (TNFR)-associated factor 6; IL-1α: interleukin 1α; IL-1β: interleukin 1β; IL-8: interleukin 8; TNF-α: tumor necrosis factor; COX-2: cyclooxygenase-2; PGE2: prostaglandin E2; LTB4: leukotriene B4; RANTES; regulated on activation, normal T cell expressed and secreted; ATG12: autophagy related 12; JNK2: c-Jun N-terminal kinases; Bcl2: B-cell lymphoma 2; CEACAM6: carcinoembryonic antigen-related cell adhesion molecule 6.
4. miR-146a as a Biomarker in Lung Cancer
4.1. Diagnostic Potential
At the time of writing this manuscript, no study had indicated miR-146a as a single diagnostic marker in lung cancer. However, its diagnostic utility has been studied along with other miRNAs in tissue or blood from lung cancer individuals. For instance, Chen X et al. observed that miR-134, miR-146a, miR-221, miR-222, and miR-23a was greatly deregulated in lung cancer and colorectal cancer sera when analyzed through Solexa sequencing and qRT-PCR [87]. Another similar study found that miR-125a-5p, miR-145, and miR-146a in serum were significantly overexpressed in NSCLC patients with miR-146a sensitivity and a respective specificity of 92.75% and 58.57% in discriminating NSCLC patients from healthy individuals [88]. Serum miR-146a upregulation along with miR-196a-2 downregulation has been significantly associated with lung cancer patients and their polymorphisms were found to be linked to lung cancer risk in Egyptian [89] and north Indian individuals [90].
4.2. Prognostic Potential
As per the meta-analysis conducted by Li et al., miR-146a upregulation showed a significant association with better overall survival in most solid tumors like lung cancer, cervical cancer, ovary cancer, liver cancer, and stomach cancer, and inhibited diverse oncogenic pathways related to phospholipase C and aquaporins [52]. Additionally, downregulation of miR-146a indicated metastases with high relapse [91] and poor overall survival in lung cancer patients [19]. Contradictory to this, serum upregulation of miR-146a indicated a successful response to chemotherapy that prolonged survival in lung cancer patients [92]. Kyong-Ah Yoon et al. found that SNPs, such as rs2910164 of miR-146a and rs11614913 of miR-196a2, were positively correlated with better relapse-free survival (RFS), especially in the advanced phase of lung cancer. Furthermore, RFS showed much better improvement in individuals with higher rs2910164 and rs11614913 SNP allele frequencies [93]. These findings were contraindicated by Wang et al., who reported significantly high levels of miR-146a in the serum of NSCLC patients in comparison to the normal controls [88]. Yan-Gang Ren conducted a meta-analysis study in which lung cancer individuals found with rs11614913 in miR-196a2 and rs2910164 in miR-146a were significantly associated with lung cancer susceptibility, which appeared to contradict the earlier studies [94]. Li et al. (2017) found a similar type of observation, which was a specific elevation of miR-146a in the serum of stage I and II lung adenocarcinoma patients [52].
This entry is adapted from the peer-reviewed paper 10.3390/diagnostics11020274
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-Small Cell Lung Cancer: Epidemiology, Risk Factors, Treatment, and Survivorship. Mayo Clin. Proc. 2008, 83, 584–594.
- Zheng, D.; Haddadin, S.; Wang, Y.; Gu, L.Q.; Perry, M.C.; Freter, C.E.; Wang, M.X. Plasma microRNAs as novel biomarkers for early detection of lung cancer. Int. J. Clin. Exp. Pathol. 2011, 4, 575–586.
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–716.
- Pammolli, F.; Magazzini, L.; Riccaboni, M. The productivity crisis in pharmaceutical R&D. Nat. Rev. Drug Discov. 2011, 10, 428–438.
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854.
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114.
- Lebanony, D.; Benjamin, H.; Gilad, S.; Ezagouri, M.; Dov, A.; Ashkenazi, K.; Gefen, N.; Izraeli, S.; Rechavi, G.; Pass, H.; et al. Diagnostic Assay Based on hsa-miR-205 Expression Distinguishes Squamous From Nonsquamous Non–Small-Cell Lung Carcinoma. J. Clin. Oncol. 2009, 27, 2030–2037.
- Landi, M.T.; Zhao, Y.; Rotunno, M.; Koshiol, J.; Liu, H.; Bergen, A.W.; Rubagotti, M.; Goldstein, A.M.; Linnoila, I.; Marincola, F.M.; et al. MicroRNA Expression Differentiates Histology and Predicts Survival of Lung Cancer. Clin. Cancer Res. 2010, 16, 430–441.
- Boeri, M.; Verri, C.; Conte, D.; Roz, L.; Modena, P.; Facchinetti, F.; Calabrò, E.; Croce, C.M.; Pastorino, U.; Sozzi, G. MicroRNA signatures in tissues and plasma predict development and prognosis of computed tomography detected lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 3713–3718.
- Gilad, S.; Lithwick-Yanai, G.; Barshack, I.; Benjamin, S.; Krivitsky, I.; Edmonston, T.B.; Bibbo, M.; Thurm, C.; Horowitz, L.; Huang, Y.; et al. Classification of the Four Main Types of Lung Cancer Using a MicroRNA-Based Diagnostic Assay. J. Mol. Diagn. 2012, 14, 510–517.
- Rosenfeld, N.; Aharonov, R.; Meiri, E.; Rosenwald, S.; Spector, Y.; Zepeniuk, M.; Benjamin, H.; Shabes, N.; Tabak, S.; Levy, A.; et al. MicroRNAs accurately identify cancer tissue origin. Nat. Biotechnol. 2008, 26, 462–469.
- Xiao, W.; Zhong, Y.; Wu, L.; Yang, D.; Ye, S.; Zhang, M. Prognostic value of microRNAs in lung cancer: A systematic review and meta-analysis. Mol. Clin. Oncol. 2018, 10, 67–77.
- Viswanathan, S.R.; Daley, G.Q. Lin28: A MicroRNA Regulator with a Macro Role. Cell 2010, 140, 445–449.
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of Tissue-Specific MicroRNAs from Mouse. Curr. Biol. 2002, 12, 735–739.
- Yin, J.; Zhao, J.; Hu, W.; Yang, G.; Yu, H.; Wang, R.; Wang, L.; Zhang, G.; Fu, W.; Dai, L.; et al. Disturbance of the let-7/LIN28 double-negative feedback loop is associated with radio- and chemo-resistance in non-small cell lung cancer. PLoS ONE 2017, 12, e0172787.
- Lagos-Quintana, M. Identification of Novel Genes Coding for Small Expressed RNAs. Science 2001, 294, 853–858.
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-B-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486.
- Chen, G.; Umelo, I.A.; Lv, S.; Teugels, E.; Fostier, K.; Kronenberger, P.; Dewaele, A.; Sadones, J.; Geers, C.; De Grève, J. miR-146a Inhibits Cell Growth, Cell Migration and Induces Apoptosis in Non-Small Cell Lung Cancer Cells. PLoS ONE 2013, 8, e60317.
- Iacona, J.R.; Monteleone, N.J.; Lutz, C.S. miR-146a suppresses 5-lipoxygenase activating protein (FLAP) expression and Leukotriene B4 production in lung cancer cells. Oncotarget 2018, 9, 26751–26769.
- Lau, N.C. An Abundant Class of Tiny RNAs with Probable Regulatory Roles in Caenorhabditis elegans. Science 2001, 294, 858–862.
- Denli, A.M.; Tops, B.B.J.; Plasterk, R.H.A.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the Microprocessor complex. Nature 2004, 432, 231–235.
- Gregory, R.I.; Yan, K.-P.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240.
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419.
- Han, J. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 18, 3016–3027.
- Zeng, Y.; Yi, R.; Cullen, B.R. Recognition and cleavage of primary microRNA precursors by the nuclear processing enzyme Drosha. Embo J. 2004, 24, 138–148.
- Han, J.; Lee, Y.; Yeom, K.-H.; Nam, J.-W.; Heo, I.; Rhee, J.-K.; Sohn, S.Y.; Cho, Y.; Zhang, B.-T.; Kim, V.N. Molecular Basis for the Recognition of Primary microRNAs by the Drosha-DGCR8 Complex. Cell 2006, 125, 887–901.
- Heo, I.; Ha, M.; Lim, J.; Yoon, M.-J.; Park, J.-E.; Kwon, S.C.; Chang, H.; Kim, V.N. Mono-Uridylation of Pre-MicroRNA as a Key Step in the Biogenesis of Group II let-7 MicroRNAs. Cell 2012, 151, 521–532.
- Burke, J.M.; Kelenis, D.P.; Kincaid, R.P.; Sullivan, C.S. A central role for the primary microRNA stem in guiding the position and efficiency of Drosha processing of a viral pri-miRNA. RNA 2014, 20, 1068–1077.
- Gregory, R.I.; Chendrimada, T.P.; Cooch, N.; Shiekhattar, R. Human RISC Couples MicroRNA Biogenesis and Posttranscriptional Gene Silencing. Cell 2005, 123, 631–640.
- Kim, Y.; Yeo, J.; Lee, J.H.; Cho, J.; Seo, D.; Kim, J.-S.; Kim, V.N. Deletion of Human tarbp2 Reveals Cellular MicroRNA Targets and Cell-Cycle Function of TRBP. Cell Rep. 2014, 9, 1061–1074.
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744.
- Liu, J.; Valencia-Sanchez, M.A.; Hannon, G.J.; Parker, R. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat. Cell Biol. 2005, 7, 719–723.
- Eulalio, A.; Behm-Ansmant, I.; Schweizer, D.; Izaurralde, E. P-Body Formation Is a Consequence, Not the Cause, of RNA-Mediated Gene Silencing. Mol. Cell. Biol. 2007, 27, 3970–3981.
- Cornett, A.L.; Lutz, C.S. Regulation of COX-2 expression by miR-146a in lung cancer cells. RNA 2014, 20, 1419–1430.
- Chang, T.-C.; Yu, D.; Lee, Y.-S.; Wentzel, E.A.; Arking, D.E.; West, K.M.; Dang, C.V.; Thomas-Tikhonenko, A.; Mendell, J.T. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat. Genet. 2007, 40, 43–50.
- Forloni, M.; Dogra, S.K.; Dong, Y.; Conte, D.; Ou, J.; Zhu, L.J.; Deng, A.; Mahalingam, M.; Green, M.R.; Wajapeyee, N. miR-146a promotes the initiation and progression of melanoma by activating Notch signaling. eLife 2014, 3, e01460.
- Kulis, M.; Esteller, M. DNA Methylation and Cancer. In Epigenetics and Cancer, Part A; Elsevier: Amsterdam, The Netherlands, 2010; pp. 27–56.
- Zhang, Z.; Zhang, Y.; Sun, X.X.; Ma, X.; Chen, Z.N. microRNA-146a inhibits cancer metastasis by downregulating VEGF through dual pathways in hepatocellular carcinoma. Mol. Cancer 2015, 14, 1–15.
- Wang, X.; Gao, H.; Ren, L.; Gu, J.; Zhang, Y.; Zhang, Y. Demethylation of the miR-146a promoter by 5-Aza-2’-deoxycytidine correlates with delayed progression of castration-resistant prostate cancer. BMC Cancer 2014, 14, 1–11.
- de Giorgio, A.; Krell, J.; Harding, V.; Stebbing, J.; Castellano, L. Emerging Roles of Competing Endogenous RNAs in Cancer: Insights from the Regulation of PTEN. Mol. Cell. Biol. 2013, 33, 3976–3982.
- Zhou, Y.-X.; Zhao, W.; Mao, L.-W.; Wang, Y.-L.; Xia, L.-Q.; Cao, M.; Shen, J.; Chen, J. Long non-coding RNA NIFK-AS1 inhibits M2 polarization of macrophages in endometrial cancer through targeting miR-146a. Int. J. Biochem. Cell Biol. 2018, 104, 25–33.
- Han, W.; Du, X.; Liu, M.; Wang, J.; Sun, L.; Li, Y. Increased expression of long non-coding RNA SNHG16 correlates with tumor progression and poor prognosis in non-small cell lung cancer. Int. J. Biol. Macromol. 2019, 121, 270–278.
- Kumaraswamy, E.; Wendt, K.L.; Augustine, L.A.; Stecklein, S.R.; Sibala, E.C.; Li, D.; Gunewardena, S.; Jensen, R.A. BRCA1 regulation of epidermal growth factor receptor (EGFR) expression in human breast cancer cells involves microRNA-146a and is critical for its tumor suppressor function. Oncogene 2014, 34, 4333–4346.
- Liu, R.; Liu, C.; Chen, D.; Yang, W.-H.; Liu, X.; Liu, C.-G.; Dugas, C.M.; Tang, F.; Zheng, P.; Liu, Y.; et al. FOXP3 Controls an miR-146/NF-κB Negative Feedback Loop That Inhibits Apoptosis in Breast Cancer Cells. Cancer Res. 2015, 75, 1703–1713.
- Sun, M.; Fang, S.; Li, W.; Li, C.; Wang, L.; Wang, F.; Wang, Y. Associations of miR-146a and miR-146b expression and clinical characteristics in papillary thyroid carcinoma. Cancer Biomark. 2015, 15, 33–40.
- Larner-Svensson, H.M.; Williams, A.E.; Tsitsiou, E.; Perry, M.M.; Jiang, X.; Chung, K.F.; Lindsay, M.A. Pharmacological studies of the mechanism and function of interleukin-1β-induced miRNA-146a expression in primary human airway smooth muscle. Respir. Res. 2010, 11, 1–13.
- Bertoli, G.; Cava, C.; Castiglioni, I. MicroRNAs: New Biomarkers for Diagnosis, Prognosis, Therapy Prediction and Therapeutic Tools for Breast Cancer. Theranostics 2015, 5, 1122–1143.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715.
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751.
- Li, M.-W.; Gao, L.; Dang, Y.-W.; Li, P.; Li, Z.-Y.; Chen, G.; Luo, D.-Z. Protective potential of miR-146a-5p and its underlying molecular mechanism in diverse cancers: A comprehensive meta-analysis and bioinformatics analysis. Cancer Cell Int. 2019, 19, 1–21.
- Pavel, A.B.; Campbell, J.D.; Liu, G.; Elashoff, D.; Dubinett, S.; Smith, K.; Whitney, D.; Lenburg, M.E.; Spira, A. Alterations in Bronchial Airway miRNA Expression for Lung Cancer Detection. Cancer Prev. Res. 2017, 10, 651–659.
- Scagliotti, G.V.; Selvaggi, G.; Novello, S.; Hirsch, F.R. The Biology of Epidermal Growth Factor Receptor in Lung Cancer. Clin. Cancer Res. 2004, 10, 4227s–4232s.
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181.
- Qi, P.; Li, Y.; Liu, X.; Jafari, F.A.; Zhang, X.; Sun, Q.; Ma, Z. Cryptotanshinone Suppresses Non-Small Cell Lung Cancer via microRNA-146a-5p/EGFR Axis. Int. J. Biol. Sci. 2019, 15, 1072–1079.
- Zucker, S.; Hymowitz, M.; Rollo, E.E.; Mann, R.; Conner, C.E.; Cao, J.; Foda, H.D.; Tompkins, D.C.; Toole, B.P. Tumorigenic Potential of Extracellular Matrix Metalloproteinase Inducer. Am. J. Pathol. 2001, 158, 1921–1928.
- Huang, W.T.; He, R.Q.; Li, X.J.; Ma, J.; Peng, Z.G.; Zhong, J.C.; Hu, X.H.; Chen, G. miR-146a-5p targets TCSF and influences cell growth and apoptosis to repress NSCLC progression. Oncol. Rep. 2019, 41, 2226–2240.
- Calandra, T.; Roger, T. Macrophage migration inhibitory factor: A regulator of innate immunity. Nat. Rev. Immunol. 2003, 3, 791–800.
- Hagemann, T.; Wilson, J.; Kulbe, H.; Li, N.F.; Leinster, D.A.; Charles, K.; Klemm, F.; Pukrop, T.; Binder, C.; Balkwill, F.R. Macrophages Induce Invasiveness of Epithelial Cancer Cells Via NF-κB and JNK. J. Immunol. 2005, 175, 1197–1205.
- Li, J.; Zhang, J.; Xie, F.; Peng, J.; Wu, X. Macrophage migration inhibitory factor promotes Warburg effect via activation of the NF-κB/HIF-1α pathway in lung cancer. Int. J. Mol. Med. 2017, 41, 1062–1068.
- Wang, W.-M.; Liu, J.-C. Effect and molecular mechanism of mir-146a on proliferation of lung cancer cells by targeting and regulating MIF gene. Asian Pac. J. Trop. Med. 2016, 9, 806–811.
- Li, Y.-L.; Wang, J.; Zhang, C.-Y.; Shen, Y.-Q.; Wang, H.-M.; Ding, L.; Gu, Y.-C.; Lou, J.-T.; Zhao, X.-T.; Ma, Z.-L.; et al. MiR-146a-5p inhibits cell proliferation and cell cycle progression in NSCLC cell lines by targeting CCND1 and CCND2. Oncotarget 2016, 7, 59287–59298.
- Shi, L.; Xu, Z.; Wu, G.; Chen, X.; Huang, Y.; Wang, Y.; Jiang, W.; Ke, B. Up-regulation of miR-146a increases the sensitivity of non-small cell lung cancer to DDP by downregulating cyclin J. BMC Cancer 2017, 17, 1–14.
- Park, D.H.; Jeon, H.S.; Lee, S.Y.; Choi, Y.Y.; Lee, H.W.; Yoon, S.; Lee, J.C.; Yoon, Y.S.; Kim, D.S.; Na, M.J.; et al. MicroRNA-146a inhibits epithelial mesenchymal transition in non-small cell lung cancer by targeting insulin receptor substrate 2. Int. J. Oncol. 2015, 47, 1545–1553.
- Heuberger, J.; Birchmeier, W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915.
- Tan, W.; Liao, Y.; Qiu, Y.; Liu, H.; Tan, D.; Wu, T.; Tang, M.; Zhang, S.; Wang, H. miRNA 146a promotes chemotherapy resistance in lung cancer cells by targeting DNA damage inducible transcript 3 (CHOP). Cancer Lett. 2018, 428, 55–68.
- Pérez-García, E.I.; Meza-Sosa, K.F.; López-Sevilla, Y.; Camacho-Concha, N.; Sánchez, N.C.; Pérez-Martínez, L.; Pedraza-Alva, G. Merlin negative regulation by miR-146a promotes cell transformation. Biochem. Biophys. Res. Commun. 2015, 468, 594–600.
- Balkwill, F.; Charles, K.A.; Mantovani, A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 2005, 7, 211–217.
- Gomes, M.; Teixeira, A.L.; Coelho, A.; Araújo, A.; Medeiros, R. The Role of Inflammation in Lung Cancer. In Advances in Experimental Medicine and Biology; Springer Basel: Basel, Switzerland, 2014; pp. 1–23.
- Richardson, C.M.; Sharma, R.A.; Cox, G.; O’Byrne, K.J. Epidermal growth factor receptors and cyclooxygenase-2 in the pathogenesis of non-small cell lung cancer: Potential targets for chemoprevention and systemic therapy. Lung Cancer 2003, 39, 1–13.
- Saba, R.; Sorensen, D.L.; Booth, S.A. MicroRNA-146a: A Dominant, Negative Regulator of the Innate Immune Response. Front. Immunol. 2014, 5, 578.
- Perry, M.M.; Moschos, S.A.; Williams, A.E.; Shepherd, N.J.; Larner-Svensson, H.M.; Lindsay, M.A. Rapid Changes in MicroRNA-146a Expression Negatively Regulate the IL-1β-Induced Inflammatory Response in Human Lung Alveolar Epithelial Cells. J. Immunol. 2008, 180, 5689–5698.
- Lambert, K.A.; Roff, A.N.; Panganiban, R.P.; Douglas, S.; Ishmael, F.T. MicroRNA-146a is induced by inflammatory stimuli in airway epithelial cells and augments the anti-inflammatory effects of glucocorticoids. PLoS ONE 2018, 13, e0205434.
- Bhaumik, D.; Scott, G.K.; Schokrpur, S.; Patil, C.K.; Orjalo, A.V.; Rodier, F.; Lithgow, G.J.; Campisi, J. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging 2009, 1, 402–411.
- Bui, N.; Woodward, B.; Johnson, A.; Husain, H. Novel Treatment Strategies for Brain Metastases in Non-small-cell Lung Cancer. Curr. Treat. Opt. Oncol. 2016, 17, 25.
- Jiang, W.G.; Sanders, A.J.; Katoh, M.; Ungefroren, H.; Gieseler, F.; Prince, M.; Thompson, S.K.; Zollo, M.; Spano, D.; Dhawan, P.; et al. Tissue invasion and metastasis: Molecular, biological and clinical perspectives. Semin. Cancer Biol. 2015, 35, S244–S275.
- Woodhouse, E.C.; Chuaqui, R.F.; Liotta, L.A. General mechanisms of metastasis. Cancer 1997, 80, 1529–1537.
- Cheung, K.J.; Ewald, A.J. A collective route to metastasis: Seeding by tumor cell clusters. Science 2016, 352, 167–169.
- Kong, W.; Yang, H.; He, L.; Zhao, J.J.; Coppola, D.; Dalton, W.S.; Cheng, J.Q. MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell. Biol. 2008, 28, 6773–6784.
- Shahriar, A.; Ghaleh-Aziz Shiva, G.; Ghader, B.; Farhad, J.; Hosein, A.; Parsa, H. The dual role of mir-146a in metastasis and disease progression. Biomed. Pharm. 2020, 126, 110099.
- Kim, J.; Yao, F.; Xiao, Z.; Sun, Y.; Ma, L. MicroRNAs and metastasis: Small RNAs play big roles. Cancer Metastasis Rev. 2018, 37, 5–15.
- Bleau, A.M.; Redrado, M.; Nistal-Villan, E.; Villalba, M.; Exposito, F.; Redin, E.; de Aberasturi, A.L.; Larzabal, L.; Freire, J.; Gomez-Roman, J.; et al. miR-146a targets c-met and abolishes colorectal cancer liver metastasis. Cancer Lett. 2018, 414, 257–267.
- Ghuwalewala, S.; Ghatak, D.; Das, S.; Das, P.; Butti, R.; Gorain, M.; Kundu, G.C.; Roychoudhury, S. MiR-146a-dependent regulation of CD24/AKT/β-catenin axis drives cancer stem cell phenotype in oral squamous cell carcinoma. bioRxiv 2019, 429068.
- Saunders, N.A.; Simpson, F.; Thompson, E.W.; Hill, M.M.; Endo-Munoz, L.; Leggatt, G.; Minchin, R.F.; Guminski, A. Role of intratumoural heterogeneity in cancer drug resistance: Molecular and clinical perspectives. Embo Mol. Med. 2012, 4, 675–684.
- Sanchez, N.C.; Medrano-Jimenez, E.; Aguilar-Leon, D.; Perez-Martinez, L.; Pedraza-Alva, G. Tumor Necrosis Factor-Induced miR-146a Upregulation Promotes Human Lung Adenocarcinoma Metastasis by Targeting Merlin. DNA Cell Biol. 2020, 39, 484–497.
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of microRNAs in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006.
- Wang, R.J.; Zheng, Y.H.; Wang, P.; Zhang, J.Z. Serum miR-125a-5p, miR-145 and miR-146a as diagnostic biomarkers in non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 765–771.
- Mohamed, R.H.; Pasha, H.F.; Gad, D.M.; Toam, M.M. miR-146a and miR-196a-2 genes polymorphisms and its circulating levels in lung cancer patients. J. Biochem. 2019, 166, 323–329.
- Sodhi, K.K.; Bahl, C.; Singh, N.; Behera, D.; Sharma, S. Functional genetic variants in pre-miR-146a and 196a2 genes are associated with risk of lung cancer in North Indians. Future Oncol. 2015, 11, 2159–2173.
- Yuwen, D.L.; Sheng, B.B.; Liu, J.; Wenyu, W.; Shu, Y.Q. MiR-146a-5p level in serum exosomes predicts therapeutic effect of cisplatin in non-small cell lung cancer. Eur. Rev. Med. Pharm. Sci 2017, 21, 2650–2658.
- Wu, C.; Cao, Y.; He, Z.; He, J.; Hu, C.; Duan, H.; Jiang, J. Serum Levels of miR-19b and miR-146a as Prognostic Biomarkers for Non-Small Cell Lung Cancer. Tohoku J. Exp. Med. 2014, 232, 85–95.
- Yoon, K.-A.; Yoon, H.; Park, S.; Jang, H.-J.; Zo, J.I.; Lee, H.-S.; Lee, J.S. The prognostic impact of microRNA sequence polymorphisms on the recurrence of patients with completely resected non–small cell lung cancer. J. Thorac. Cardiovasc. Surg. 2012, 144, 794–807.
- Ren, Y.-G.; Zhou, X.-M.; Cui, Z.-G.; Hou, G. Effects of common polymorphisms in miR-146a and miR-196a2 on lung cancer susceptibility: A meta-analysis. J. Thorac. Dis. 2016, 8, 1297–1305.