The production of melanin pigments by melanocytes and their quantity, quality, and distribution play a decisive role in determining human skin, eye, and hair color, and protect the skin from adverse effects of ultraviolet radiation (UVR) and oxidative stress from various environmental pollutants. Melanocytes reside in the basal layer of the interfollicular epidermis and are compensated by melanocyte stem cells in the follicular bulge area. Various stimuli such as eczema, microbial infection, ultraviolet light exposure, mechanical injury, and aging provoke skin inflammation. These acute or chronic inflammatory responses cause inflammatory cytokine production from epidermal keratinocytes as well as dermal fibroblasts and other cells, which in turn stimulate melanocytes, often resulting in skin pigmentation. It is confirmed by some recent studies that several interleukins (ILs) and other inflammatory mediators modulate the proliferation and differentiation of human epidermal melanocytes and also promote or inhibit expression of melanogenesis-related gene expression directly or indirectly, thereby participating in regulation of skin pigmentation. Understanding of mechanisms of skin pigmentation due to inflammation helps to elucidate the relationship between inflammation and skin pigmentation regulation and can guide development of new therapeutic pathways for treating pigmented dermatosis.
- melanogenesis
- skin pigmentation
- inflammation
- inflammatory cytokine
1.Skin Pigmentation
Skin color is one of the topmost concerns for human beings as it provides uniformity, identity and represents the overall appearance of a person [1]. Production, quantity, quality, and distribution of melanin, a group of natural pigments, determines the color of human skin, eyes, and hair. It is crucially involved in the defense mechanism to protect skin from adverse effects of ultraviolet radiation (UVR) and oxidative stress of environmental pollutants [2]. Synthesis of melanin starts with formation of melanosomes, melanin-containing organelles, in epidermal melanocytes by a process called melanogenesis [3][4]. Melanocytes are specialized cells derived from unpigmented precursor cells called melanoblasts, originating from embryonic neural crest cells which can migrate towards the skin and other tissues during embryogenesis [5][6][7][8]. Skin pigmentation is a specific and complex mechanism that occurs due to accumulation of melanosomes in keratinocytes to protect skin from solar irradiation. Melanin pigment can be divided into two types—pheomelanin and eumelanin. Pheomelanin has a reddish to brownish color, causing reactive oxygen species production under UVR stimulation. Eumelanin has black color, which works mainly to protect the nucleus from UVR. Skin color differences can emerge from the quantity and quality (pheo/eumelanin ratio) of melanin produced as well as size, number, composition, mode of transfer, distribution, and degradation of melanosomes inside keratinocytes, whereas melanocytes numbers typically remain relatively constant [8]. Keratinocytes have a significant role in regulating adhesion, proliferation, survival, and morphology of melanocytes [8].
There are various nonspecific intrinsic factors (hormonal environment, inflammation) and extrinsic factors (solar irradiation such as ultraviolet irradiation, environmental pollution, drugs) modulating genetically determined melanin levels. Various stimuli including skin eruptions such as eczema, allergens, microbial infection, pathogens, chemical stimuli and physical damage, ultraviolet light exposure, and aging can all lead to skin inflammation [9][10][11][12], defined as a defensive reaction of living tissues with a vascular system in response to exogenous and endogenous stimuli [13]. In dermatology and cosmetology, hyperpigmentation or hypopigmentation after inflammation is a major problem and commonly seen due to acute or chronic inflammatory skin reactions [14]. Inflammatory mediators are chemical factors involved in mediating inflammatory reactions and mainly secreted by T helper cells (Th), monocytes and macrophages, dendritic cells, as well as epidermal keratinocytes. Recent studies have revealed a close relationship between inflammatory cytokines and skin pigmentation. Diversified interleukins (ILs) and varieties of inflammatory mediators can also participate in melanogenesis regulation by modulating proliferation and differentiation of human epidermal melanocytes, and also promote or inhibit melanogenesis-related gene expression directly or indirectly [14][15][16]. Many genes are involved in regulating pigmentation at various levels, as well as mutations in several cause pigmentary disorders.
2. Melanogenesis Regulation
MITF is a basic helix–loop–helix leucine zipper (bHLH-ZIP) transcription factor belonging to the MIT regulating melanocyte function. MITF controls expression of the melanogenesis enzymes TYR, TYRP1 and TYRP2 [17] and regulates melanocyte functions, including pigmentation, melanocyte differentiation, proliferation, and cell survival [18]. Furthermore, MITF protein plays a vital role in expression regulation of the RAB27A protein, crucial in melanosome transport and the melanosomal matrix protein Pmel17 [19][20][21][22]. It has also been discovered that the MIT (microphthalmia) family is the main transcriptional regulator of many genes coding for proteins involved in pigment synthesis, melanosomal structure, as well as melanosome trafficking [17]. Many other transcription factors are involved in regulating MITF expression, including a paired box protein 3 (PAX3), sex-determining region Y-box 9 and 10 (SOX9, SOX10), lymphoid enhancer-binding factor 1 (LEF-1) and Cyclic adenosine monophosphate (cAMP) responsive-element binding protein (CREB), which is phosphorylated by signals via melanocortin-1 receptor (MC1R) [23]. Recent findings have elucidated MITF expression regulation in mice melanocytes is influenced by salt-inducible kinase (SIK), which phosphorylates CREB-regulated transcription coactivator (CRTC) to prevent its nuclear translocation, resulting in inhibited CREB-induced MITF upregulation [24].
Involvement of Signaling Pathways in Melanogenesis Regulation
The regulation of melanogenesis is a complex process and more than 150 genes are involved in this regulation [25]. Melanogenesis can be controlled at different levels as many of these genes affect the developmental process crucial to melanoblasts, while others are involved in melanocyte survival and differentiation [26]. At least 25 of these genes are involved in regulating different pigmentation pathways or function of melanosomes. The cAMP/protein kinase A (PKA) signaling pathway is one of the most significant signaling pathways and the most well-known receptor is the MC1R. MC1R belongs to G protein-coupled receptor family and is expressed predominantly in melanocytes. The α-melanocyte-stimulating hormone (α-MSH) binds to MC1R and activates adenylate cyclase (AC), increasing intracellular concentration of secondary messenger, cAMP. The cAMP activates PKA, then PKA phosphorylates CREB, leading to increased expression of MITF, resulting in melanogenesis promotion [19][27][28][29].
In addition to the above, other signaling pathways such as mitogen-activated protein kinase (MAPK), inositol trisphosphate/diacylglycerol (IP3/DAG), wingless-type protein (WNT), and PKC are reported to participate in the melanogenesis process. The IP3/DAG pathway is activated by the α1 adrenergic receptor (α1AR) and thus increases the intracellular levels of PKC-β and activates tyrosinase [30]. The c-KIT, granulocyte-macrophage colony-stimulating factor receptor (GM-CSFR), and HGF receptor-mediated signaling pathways can be activated by SCF, GM-CSF and HGF, respectively, which leads to autophosphorylation and activation of MAPK, thereby phosphorylating MITF and upregulating melanogenesis-related enzyme expression [3][31][32]. The WNT bind to class frizzled (FZD) receptors and the wingless-type protein (WNT)/FZD signaling pathway is involved in several aspects of the mouse melanocyte physiology, from developmental lineage differentiation to subsequent maintenance of melanocytes [33]. Indeed, pigment development and differentiation from the neural crest cells into melanocytes can be promoted by WNT1 and WNT3A in zebrafish [34]. Several models including mouse Melan-a cells and avian embryo melanocytes have demonstrated WNT1 is responsible for increasing the number of melanoblasts differentiating from melanoblast precursors, whereas WNT3A is involved in promotion of the differentiation of neural crest cells and melanoblasts into melanocytes by maintaining MITF expression [35][36][37]. In general, the WNT signaling pathway can activate the melanocytes-specific MITF isoform (MITF-M) promoter [38][39][40] and upregulate MITF expression, thus participating in melanogenesis regulation. Catecholamines can promote melanogenesis through the cAMP/PKA pathway and also mediate melanogenesis through PKC-β pathways by α1 and β2 adrenergic receptors [3][30].
3. Inflammatory Cytokines
Allergens, pathogens, chemical stimuli, physical damage, and UVR can all lead to skin inflammation and inflammatory cytokines are predominantly released from immune cells, including monocytes, macrophages, and lymphocytes, which are considered closely related to skin pigmentation [41][42]. Pro- and anti-inflammatory cytokines facilitate and inhibit inflammation. Cytokines modulate immune response to infection or inflammation and regulate inflammation itself via a complex network of interactions. Th1, Th2 and Th17 cells [43] are the main types of Th cells in inflammation. Th1 cells secret cytokines such as interferon-γ (IFN-γ), TNF, and IL-2 and thus play an important role in cellular immune responses and Th2 cells play a central role in humoral immune responses by secreting IL-4, IL-5, IL-10, and IL-13 as well as other cytokines [44][45]. Th17 cells are a recently recognized subpopulation of T cells and produce IL-17, and IL-22, and IFN-γ. Skin samples from atopic dermatitis (AD) patients contain numerous Th17 and Th2 cells, and cytokines produced from these cells (especially Th2 cells), which reduce the epidermis barrier function, elevate trans-epidermal water loss, and increase permeation of environmental irritants and allergens. Antigen-presenting cells (APCs) in the epidermis and Langerhans cells capture allergens with their dendrites extending upwards and process and present them to lymphocytes in the draining lymph nodes, thus stimulate naïve T-cells to be effector memory T cells.
Exposure to sensitized allergens evokes activation and accumulation of effector memory T cells to the exposure site, and these cells are stimulated to produce proinflammatory cytokines into the inflamed area. Atopic dermatitis patients are usually sensitized to food allergens, such as egg, and wheat, or to environmental allergens, such as house dust mites and pollen. These allergens existing everywhere can easily provoke inflammation in barrier-disrupted, easily permeable skin of atopic dermatitis patients. Atopic dirty neck is a well-known pigmentary condition considered to be due to post-inflammatory pigmentation/depigmentation. Th17 secretes IL-17, IL-6, IL-21, and IL-22, which participate in innate and acquired immunity. Psoriasis is another major inflammatory skin disease, where Th17 cells play an inevitable role in its pathogenesis. Treatment with biologics often leaves hypopigmented/hyperpigmented macules in the place of psoriasis plaques, which may be reflected by cytokine profiles in the psoriasis plaques. Treatment with UV (Psolalen-UVA (PUVA) or narrowband UVB) leaves strongly hyperpigmented macules without exception, probably due to the melanogenic function of UV.
4. Inflammatory Cytokines Induce Melanogenesis
Several studies have observed the involvement of local inflammatory factors in skin pigmentation regulation, which are presented in Table 1 (Figure 3). It has been revealed that IL-4 directly takes part in inhibiting melanogenesis in normal human epidermal melanocytes (NHEMs) by downregulating MITF, TYRP1, and TYRP2 expression through the JAK2-STAT6 signaling pathway [16]. IL-4 is mainly produced by Th2 cells. In chronic inflammation, basophils, eosinophils, and mast cells can also secrete IL-4 [46][47]. IL-4 plays a crucial role in IgE generation in hypersensitivity, inflammation induction, autoimmunity [48] and is also involved in the maintenance of Th2 lymphocytes and acts as an autocrine growth factor of differentiated Th2 cells [49]. It is hypothesized that skewed balance to Th1 is directly responsible for vitiligo development by changing IFN-γ/IL-4, Tbet/Gata3 profiles in vitiligo patients compared to controls [50][51].
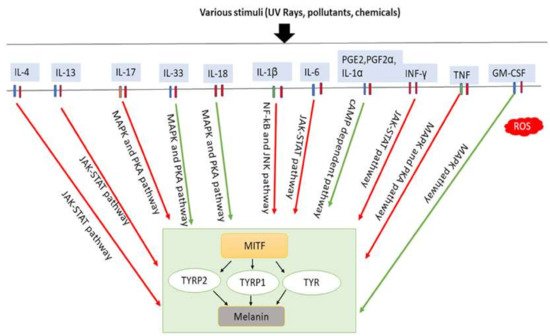
Figure 3. Involvement of inflammatory factors in melanogenesis. Inflammatory factors including IL-33, IL-18, and GM-CSF promote melanogenesis by stimulating the MAPK and PKA pathways either simultaneously or individually. In addition, PGE2, PGF2α, and IL-1α promote melanogenesis by stimulating cAMP-dependent pathway. In contrast, inflammatory factors such as IL-4, IL-13, IL-6, IL-17, IL-1β, TNF and IFN-γ inhibit melanogenesis by suppressing the PKA and MAPK, or JAK-STAT, or NF-κβ and JNK pathways. IL-33: interleukin-33; IL-18: interleukin-18; PGE2: prostaglandin E2; PGF2α: prostaglandin F2α; IL-1α: interleukin-1α; GM-CSF: granulocyte-macrophage colony stimulating factor; IL-4: interleukin-4; IL-13: interleukin-13; IL-6: interleukin-6; IL-17: interleukin-17; IL-1β: interleukin-1β; TNF: tumor necrosis factor; IFN-γ: interferons-γ; PKA: protein kinase A; MAPK: mitogen-activated protein kinase; JNK: c-Jun N-terminal kinases; JAK-STAT: Janus kinase-signal transducer and activator of transcription; NF-κβ: nuclear factor kappa-B; TYR: tyrosinase; TRRP1: tyrosinase-related protein-1; TRRP2: tyrosinase-related protein-2. Red arrows indicates inhibition; green arrows indicate promotion.
IL-13 is a typical Th2 cell cytokine also secreted by CD4 cells, natural killer T cells, mast cells, basophils, eosinophils, and group 2 innate lymphoid cells (nuocytes) [52] sharing a receptor of similar structure and JAK2-STAT6 signaling pathway with IL-4 [53][54]. Recently, it has been found that activated human epidermal γδ T cells produce larger amounts of IL-13 than IL-4 [55] and enhance IL-13 production following treatment with Ginsenoside F1 (GF1) extracted from plant genus Panax. This plays a role in the skin-whitening effect of GF1 via effective suppression of tyrosinase and DCT, indicating that IL-13 could be a direct modulator of melanogenesis through the JAK2-STAT6 signaling pathway in skin disorders accompanied by enhanced T-cell responses, such as atopic dermatitis and vitiligo [56].
IL-17 is known to prevent bacterial and fungal infections [57] and can induce the release of large amounts of proinflammatory cytokines from a variety of cells, such as epithelial cells, endothelial cells, and fibroblasts [58], and its effects are vastly increased when cooperating with TNF [59]. IL-17, a proinflammatory cytokine, is produced primarily by Th17 cells, as well as by other immune cells, including neutrophils, natural killer cells, mast cells, αβ and γδT cells, and group 3 innate lymphoid cells. Studies have revealed IL-17 can synergize with TNF to inhibit signaling pathway for melanogenesis, thereby inhibiting pigmentation [42]. For example, studies on psoriasis lesions showed that overexpression of IL-17 and TNF increase melanocyte number and cause a simultaneous decrease in pigmentation signaling. Neutralization of TNF and/or IL-17 with mAbs revealed that both can recover growth and pigment production of melanocytes, which may contribute to pigmentation changes associated with psoriasis treatment [42].
| Cytokines | Main Source | Cells Used for Experiment | Impact on Melanogenesis | Mechanisms of Melanogenesis | Refs. |
|---|---|---|---|---|---|
| IL-4 | Th-cells | Melanocytes | Inhibition | JAK2-STAT6 signaling pathway is used for downregulating the expression of MITF, TYRP1, TYRP2 | [16] |
| IL-13 | Th2 | γδ T-cell | Inhibition | Suppress tyrosinase and DCT through the JAK2–STAT6 | [56] |
| IL-17 | Epithelial cells, Endothelial cells, Fibroblasts |
Melanocytes, Primary pooled human keratinocytes | Inhibition | TNF and IL-17 simultaneously inhibit melanin formation through PKA and MAPK signaling pathways | [42] |
| IL-33 | Keratinocytes, Fibroblasts | Keratinocytes | Promotion | Activate p38 and PKA pathways and thus promote MITF, TYR, TYRP1, TYRP2 expression | [60][61] |
| IL-18 | Monocytes/Macrophages, Keratinocytes | Melanocytes | Promotion | Increase the activity of tyrosinase and upregulate the expression Of TYRP1 and TYRP2 | [62] |
| IL-1α | Langerhans cells, Melanocytes, keratinocytes | Primary melanocytes and swine skin | Promotion | Melanin deposition is increased when IL-1α is combined with KGF | [63] |
| IL-1β | Monocytes/Macrophages, Keratinocytes | Melanoma cell lines (LB2259-MEL and CP50-MEL) | Inhibition | Through the NF-κB and JNK pathways, it downregulates MITF-M expression | [64] |
| IL-6 | Macrophages, T-cells, Adipocyte | Melanocytes | Inhibition | Tyrosinase activity is declined | [15] |
| PGE2 and PGF2α | Fibroblasts, Keratinocytes | Keratinocytes | Promotion | Activate cAMP-dependent pathway and stimulate melanocyte dendrite formation | [65] |
| INF-γ | T-cells, NK cells, NKT cells | B16F10 | Inhibition | Block melanosomal maturation and upregulate STAT1 phosphorylation | [66][67] |
| TNF | Monocytes, Macrophages, Keratinocytes, Dendritic cells, Th1, Th17 and Th22 | Melanocytes, Primary pooled human keratinocytes |
Promotion | Stimulate PKA and MAPK signaling pathways with combination of IL17, and thus inhibit melanin formation | [42] |
| GM-CSF | Macrophages, Keratinocytes and Th cells | Melanocytes | Promotion | Stimulate MAPK pathway and promote melanocyte proliferation, melanin and synthesis | [68] |
Recent research has shown that IL-33 can improve melanin biosynthesis in NHEM, stimulate phosphorylation of p38 and CREB, and increase TYR, TYRP1 and TYRP2 (DCT) expression through MITF, resulting in augmentation of melanogenesis [61]. Abundant IL-33 mRNA is found in keratinocytes induced by UVB [60] and fibroblasts [69][70], suggesting injury on skin causes IL-33 release, resulting in skin pigmentation. Initially, IL-33 can induce mast cells to produce proinflammatory cytokines and chemokines (including monocyte chemoattractant protein-1 and macrophage inflammatory protein-1α) [71][72][73][74][75], leading to activation of macrophages [76][77][78], CD 4+T cells, basophils, dendritic cells and neutrophils [71][78][79][80][81], and likely promoting Th2-skewed skin inflammation, which is another indirect effect on pigmentation by IL-33 [82]. It has also been discovered that IL-33 activates and binds its receptor suppressing tumorigenicity 2 (ST2)L/IL-1RAcP on different types of cells, including mast cells and Th2 cells [83]. Then, IL-33 activates NF-κB, suggesting that it regulates outcome of diseases such as atopic dermatitis, which can trigger pigmentation [83].
IL-18 may also modulate both innate and adaptive immunity and its dysregulation can cause autoimmune or inflammatory disease, but some studies recently suggest its participation in pigmentation regulation. It has been observed that IL-18 promotes melanogenesis and upregulates TYRP1 and TYRP2 expression [62][67] by activating pathways and increasing cascade expressing MITF [62][67]. These results indicate the involvement of IL-18 in the regulation of pigmentation by regulating melanogenesis.
IL-1 is an important proinflammatory cytokine involved in initiating inflammation and immunological reactions [84], which contributes to increased tumor invasiveness, metastasis, and angiogenesis under chronic inflammatory conditions [64]. IL-1 is produced predominantly from monocytes and macrophages [85], although it is also produced by another types of cells, such as keratinocytes and fibroblasts [86]. IL-1 possesses two forms, IL-1α and IL-1β [87]. Although they share only 24% identity in protein sequence, IL-1β and IL-1α fold in an extremely similar manner and bind to the same receptor—the type I and type II IL-1 receptor (IL-1RI, IL-1RII, respectively) [88]. The expression patterns of IL-1α and IL-1β vary among cell types [89]. IL-1α is predominantly produced by keratinocytes. Keratinocytes secrete both IL-1α and IL-1β molecules in vitro [86], in addition to the expression of IL-1RI, IL-1RII [90]. The signal transduction is initiated by binding to IL-1RI [91] which can inhibit tyrosinase activity and melanogenesis [15][64] and also stimulates human fibroblasts to produce keratinocyte growth factor (KGF) [92]. IL-1α released from keratinocytes has been shown to regulate the homeostasis of mammalian skin [93] and sufficient to trigger skin inflammation in humans [90][93] and produce more IL-1α upon UVB exposure [94]. It is documented that KGF-induced TYR expression in primary melanocytes [63]. The combination of KGF and IL-1α increases melanin deposition and is responsible for initial stage of human solar lentigines [63]. On the other hand, it was observed that the expression of MITF-M was inhibited after IL-1β treatment to a panel of melanoma cell lines. The inhibitory effects of IL-1β on melanogenesis were removed by inactivation of NF-κB and JNK pathways, suggesting IL-1β could inhibit melanogenesis by downregulating MITF-M through NF-kB and JNK pathways [64].
IL-6 is produced by numerous different cell types including keratinocytes, fibroblasts, and dermal endothelial cells and plays a critical role in regulating acute phase response, hematopoiesis, inflammation, metabolic control, liver regeneration, bone metabolism and cancer progression by promoting cell growth, survival, and differentiation [95]. It has been revealed in recent studies that the IL-6 cytokine elicits a dose-dependent decrease in tyrosinase activity, thus inhibiting melanocyte proliferation and melanogenesis [15].
PGE2 is the most abundant prostaglandin released by keratinocytes in response to UVR and inflammatory conditions such as wound healing and stimulates the formation of dendrites in melanocytes [96][97]. A wide range of physiological processes in the skin, including immune function, carcinogenesis, cutaneous barrier function, cell growth, and differentiation are regulated by PGE2 [98][99][100][101]. Biosynthesis of PGE2 is controlled by phospholipase A2 (PLA2), cyclooxygenase (COX), and prostaglandin E synthase (PGES) enzymes. PGF2α is produced by fibroblasts and keratinocytes and stimulates melanocyte dendrite formation and tyrosinase activation, but not proliferation [100]. PGE2 binds to four distinct G-protein coupled receptors (EP1–4) and EP4 receptor signaling stimulates cAMP production in melanocytes. EP3 receptor-lowered basal cAMP levels suggest relative levels of activity of these receptors’ control effects of PGE2 on cAMP in melanocytes that stimulates tyrosinase activity and proliferation [65][100].
IFN-γ is also a common secretory cytokine in the skin and the most important endogenous mediator of immunity and inflammation [67]. Th1 lymphocytes, CD 8+ cytotoxic T lymphocytes and NK cells mainly release the proinflammatory cytokine IFN-γ [102]. However, IFN-γ is also secreted by other cells, including antigen-presenting cells, B cells and NKT cells [103][104][105]. A clear relationship between increased IFN-γ signaling and a decreased number of stage III and IV melanosomes in the lesional skin of leprosy patients have been shown [66]. Specific IRF1 binding sites are in the promoter region of pigmentation-related genes such as TYR, TYRP2 (DCT). It is shown that IFN-γ negatively regulates the expression of DCT and other pigmentation-related genes, providing strong evidence that IFN-γ had a significant role in downregulating melanosome maturation [66] —i.e., skin hypopigmentation [66]. Two other crucial mediators of skin pigmentation, α-MSH and TGF-β, are involved in the protective tanning response and maintenance of melanocytes in an immature state [106][107] and are released during inflammation or UV exposure. Thus, Natarajan VT et al. proposed an αβγ-cytokine regulatory model of melanogenesis [66]. Recent studies in various mouse models of vitiligo have demonstrated that local IFN-γ accumulation produced by melanocyte-specific CD 8+ T cells plays an important role in skin depigmented spots [108][109] and also revealed increased IFN-γ is essential for vitiligo pathogenesis by inducing apoptosis of melanocytes [110]. Moreover, IFN-γ can upregulate STAT1 phosphorylation, and JAK1 inhibitors can suppress inhibiting effect of IFN-γ, thus contributing to melanogenesis. Other studies have also shown that IFN-γ inhibits IL-18-induced melanogenesis [67].
TNF functions by binding to two different receptors, TNFR1/p55 and TNFR2/p75 [41], and is secreted mainly by monocytes and macrophages, and also by keratinocytes, dendritic cells, Th1, Th17 and Th22. TNF induces inflammation through activation of vascular endothelial cells and immune cells and plays a crucial role as a regulator of lymphoid tissue development by controlling apoptosis [41]. In several autoimmune diseases, increased levels of TNF have been found at inflammation sites, such as in the lesional skin of psoriasis patients [111]. Overexpression of IL-17 and TNF synergistically modulate cytokine expression, increase melanocyte number while suppressing pigmentation signaling [42]. Therapeutic neutralization of TNF and IL-17 with mAbs resulted in a rapid recovery of pigmentation-related gene expression in psoriasis lesions, indicating IL-17 and TNF can affect pigment production in melanocytes, which may contribute to the hypopigmentation associated with psoriasis. For example, after 24–48h treatment with both IL-17 and TNF of melanocytes, decreased levels of c-KIT, MC1R, MITF, and TYRP2 were observed [42]. Consequently, the levels of tyrosinase and melanin were significantly reduced [42]. It has been suggested the combination of IL-17 and TNF can inhibit melanogenesis through PKA and MAPK signaling pathways [41][42] and blocking TNF can lead to rapid restoration of pigmentation gene expression in psoriatic lesions, indicating that anti-TNF has potential for treating depigmented dermatosis [42]. IL-33 expression is induced by IL-17 and IFN-γ in epidermal keratinocytes and induces melanogenesis, which creates a negative feedback loop in epidermal pigmentation [112][113].
GM-CSF may be a vital factor in the melanogenesis of the skin because of its role in promoting melanocyte proliferation and melanin synthesis. GM-CSF is produced by mononuclear macrophages, keratinocytes and Th cells [68]. It has been found GM-CSF synthesis and secretion by HaCaT cells increased cell proliferation in the wound healing process and contributes to wound healing and melanogenesis at the site of injury [114]. In addition, to predict the prognosis of transplantation of cultured autologous melanocytes (TCAM) in vitiligo patients, increased serum levels of GM-CSF could be used as the serum biomarkers [115].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22083970
References
- Chuong, C.M.; Nickoloff, B.J.; Elias, P.M.; Goldsmith, L.A.; Macher, E.; Maderson, P.A.; Sundberg, J.P.; Tagami, H.; Plonka, P.M.; Thestrup-Pederson, K.; et al. What is the ‘true’ function of skin? Exp. Dermatol. 2002, 11, 159–187.
- Brenner, M.; Hearing, V.J. The protective role of melanin against UV damage in human skin. Photochem. Photobiol. 2008, 84, 539–549.
- Bonaventure, J.; Domingues, M.J.; Larue, L. Cellular and molecular mechanisms controlling the migration of melanocytes and melanoma cells. Pigment. Cell Melanoma Res. 2013, 26, 316–325.
- Jan Borovansky, P.A.R. Melanins and Melanosomes Biosynthesis, Biogenesis, Physiological, and Pathological Functions; Jan Borovansky, P.A.R., Ed.; Wiley-Blackwell: Weinheim, Germany, 2011; p. 429.
- Lei, T.C.; Virador, V.; Yasumoto, K.; Vieira, W.D.; Toyofuku, K.; Hearing, V.J. Stimulation of melanoblast pigmentation by 8-methoxypsoralen:the involvement of microphthalmia-associated transcription factor, the protein kinase a signal pathway, and proteasome-mediated degradation. J. Investig. Dermatol. 2002, 119, 1341–1349.
- Sviderskaya, E.V.; Hill, S.P.; Balachandar, D.; Barsh, G.S.; Bennett, D.C. Agouti signaling protein and other factors modulating differentiation and proliferation of immortal melanoblasts. Dev. Dyn. 2001, 221, 373–379.
- Tolleson, W.H. Human melanocyte biology, toxicology, and pathology. J. Environ. Sci. Health C Environ. Carcinog Ecotoxicol. Rev. 2005, 23, 105–161.
- Lin, J.Y.; Fisher, D.E. Melanocyte biology and skin pigmentation. Nature 2007, 445, 843–850.
- Martin, S.F. Contact dermatitis: From pathomechanisms to immunotoxicology. Exp. Dermatol. 2012, 21, 382–389.
- Miller, L.S.; Cho, J.S. Immunity against Staphylococcus aureus cutaneous infections. Nat. Rev. Immunol. 2011, 11, 505–518.
- Behrends, U.; Peter, R.U.; Hintermeier-Knabe, R.; Eissner, G.; Holler, E.; Bornkamm, G.W.; Caughman, S.W.; Degitz, K. Ionizing radiation induces human intercellular adhesion molecule-1 in vitro. J. Investig. Dermatol. 1994, 103, 726–730.
- Fuchs, J.; Kern, H. Modulation of UV-light-induced skin inflammation by D-alpha-tocopherol and L-ascorbic acid: A clinical study using solar simulated radiation. Free Radic. Biol. Med. 1998, 25, 1006–1012.
- Bäsler, K.; Brandner, J.M. Tight junctions in skin inflammation. Pflugers Arch. 2017, 469, 3–14.
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228.
- Swope, V.B.; Abdel-Malek, Z.; Kassem, L.M.; Nordlund, J.J. Interleukins 1 alpha and 6 and tumor necrosis factor-alpha are paracrine inhibitors of human melanocyte proliferation and melanogenesis. J. Investig. Dermatol. 1991, 96, 180–185.
- Choi, H.; Choi, H.; Han, J.; Jin, S.H.; Park, J.Y.; Shin, D.W.; Lee, T.R.; Kim, K.; Lee, A.Y.; Noh, M. IL-4 inhibits the melanogenesis of normal human melanocytes through the JAK2-STAT6 signaling pathway. J. Investig. Dermatol. 2013, 133, 528–536.
- Vachtenheim, J.; Borovanský, J. “Transcription physiology” of pigment formation in melanocytes: Central role of MITF. Exp. Dermatol. 2010, 19, 617–627.
- Liu, J.J.; Fisher, D.E. Lighting a path to pigmentation: Mechanisms of MITF induction by UV. Pigment. Cell Melanoma Res. 2010, 23, 741–745.
- Park, H.Y.; Kosmadaki, M.; Yaar, M.; Gilchrest, B.A. Cellular mechanisms regulating human melanogenesis. Cell Mol. Life Sci. 2009, 66, 1493–1506.
- Dessinioti, C.; Stratigos, A.J.; Rigopoulos, D.; Katsambas, A.D. A review of genetic disorders of hypopigmentation: Lessons learned from the biology of melanocytes. Exp. Dermatol. 2009, 18, 741–749.
- Park, H.Y. Biology of melanocytes. In Fitzpatrick’s Dermatology in General Medicine; McGraw-Hill: New York, NY, USA, 2008; pp. 591–608.
- Steingrímsson, E.; Copeland, N.G.; Jenkins, N.A. Melanocytes and the microphthalmia transcription factor network. Annu. Rev. Genet. 2004, 38, 365–411.
- Yamaguchi, Y.; Hearing, V.J. Physiological factors that regulate skin pigmentation. Biofactors 2009, 35, 193–199.
- Mujahid, N.; Liang, Y.; Murakami, R.; Choi, H.G.; Dobry, A.S.; Wang, J.; Suita, Y.; Weng, Q.Y.; Allouche, J.; Kemeny, L.V.; et al. A UV-Independent Topical Small-Molecule Approach for Melanin Production in Human Skin. Cell Rep. 2017, 19, 2177–2184.
- Yamaguchi, Y.; Brenner, M.; Hearing, V.J. The regulation of skin pigmentation. J. Biol. Chem. 2007, 282, 27557–27561.
- Bennett, D.C.; Lamoreux, M.L. The color loci of mice—a genetic century. Pigment. Cell Res. 2003, 16, 333–344.
- Schiaffino, M.V. Signaling pathways in melanosome biogenesis and pathology. Int. J. Biochem. Cell Biol. 2010, 42, 1094–1104.
- Yuan, X.H.; Jin, Z.H. Paracrine regulation of melanogenesis. Br. J. Dermatol. 2018, 178, 632–639.
- Swope, V.B.; Abdel-Malek, Z.A. MC1R: Front and Center in the Bright Side of Dark Eumelanin and DNA Repair. Int. J. Mol. Sci. 2018, 19.
- Grando, S.A.; Pittelkow, M.R.; Schallreuter, K.U. Adrenergic and cholinergic control in the biology of epidermis: Physiological and clinical significance. J. Investig. Dermatol. 2006, 126, 1948–1965.
- Besmer, P.; Murphy, J.E.; George, P.C.; Qiu, F.H.; Bergold, P.J.; Lederman, L.; Snyder, H.W., Jr.; Brodeur, D.; Zuckerman, E.E.; Hardy, W.D. A new acute transforming feline retrovirus and relationship of its oncogene v-kit with the protein kinase gene family. Nature 1986, 320, 415–421.
- Yarden, Y.; Kuang, W.J.; Yang-Feng, T.; Coussens, L.; Munemitsu, S.; Dull, T.J.; Chen, E.; Schlessinger, J.; Francke, U.; Ullrich, A. Human proto-oncogene c-kit: A new cell surface receptor tyrosine kinase for an unidentified ligand. Embo J. 1987, 6, 3341–3351.
- Lim, X.; Nusse, R. Wnt signaling in skin development, homeostasis, and disease. Cold Spring Harb. Perspect. Biol. 2013, 5.
- Dorsky, R.I.; Moon, R.T.; Raible, D.W. Control of neural crest cell fate by the Wnt signalling pathway. Nature 1998, 396, 370–373.
- Takeda, K.; Yasumoto, K.; Takada, R.; Takada, S.; Watanabe, K.; Udono, T.; Saito, H.; Takahashi, K.; Shibahara, S. Induction of melanocyte-specific microphthalmia-associated transcription factor by Wnt-3a. J. Biol. Chem. 2000, 275, 14013–14016.
- Jin, E.J.; Erickson, C.A.; Takada, S.; Burrus, L.W. Wnt and BMP signaling govern lineage segregation of melanocytes in the avian embryo. Dev. Biol. 2001, 233, 22–37.
- Dunn, K.J.; Brady, M.; Ochsenbauer-Jambor, C.; Snyder, S.; Incao, A.; Pavan, W.J. WNT1 and WNT3a promote expansion of melanocytes through distinct modes of action. Pigment. Cell Res. 2005, 18, 167–180.
- Dorsky, R.I.; Raible, D.W.; Moon, R.T. Direct regulation of nacre, a zebrafish MITF homolog required for pigment cell formation, by the Wnt pathway. Genes Dev. 2000, 14, 158–162.
- Flaherty, K.T.; Hodi, F.S.; Fisher, D.E. From genes to drugs: Targeted strategies for melanoma. Nat. Rev. Cancer 2012, 12, 349–361.
- Widlund, H.R.; Horstmann, M.A.; Price, E.R.; Cui, J.; Lessnick, S.L.; Wu, M.; He, X.; Fisher, D.E. Beta-catenin-induced melanoma growth requires the downstream target Microphthalmia-associated transcription factor. J. Cell Biol. 2002, 158, 1079–1087.
- Grine, L.; Dejager, L.; Libert, C.; Vandenbroucke, R.E. An. inflammatory triangle in psoriasis: TNF, type I IFNs and IL-17. Cytokine Growth Factor Rev. 2015, 26, 25–33.
- Wang, C.Q.F.; Akalu, Y.T.; Suarez-Farinas, M.; Gonzalez, J.; Mitsui, H.; Lowes, M.A.; Orlow, S.J.; Manga, P.; Krueger, J.G. IL-17 and TNF synergistically modulate cytokine expression while suppressing melanogenesis: Potential relevance to psoriasis. J. Investig. Dermatol. 2013, 133, 2741–2752.
- Mosmann, T.R.; Sad, S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol. Today 1996, 17, 138–146.
- O’Garra, A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity 1998, 8, 275–283.
- Reiner, S.L.; Seder, R.A. Dealing from the evolutionary pawnshop: How lymphocytes make decisions. Immunity 1999, 11, 1–10.
- Barata, L.T.; Ying, S.; Meng, Q.; Barkans, J.; Rajakulasingam, K.; Durham, S.R.; Kay, A.B. IL-4- and IL-5-positive T lymphocytes, eosinophils, and mast cells in allergen-induced late-phase cutaneous reactions in atopic subjects. J. Allergy Clin. Immunol. 1998, 101, 222–230.
- Min, B.; Prout, M.; Hu-Li, J.; Zhu, J.; Jankovic, D.; Morgan, E.S.; Urban, J.F., Jr.; Dvorak, A.M.; Finkelman, F.D.; LeGros, G.; et al. Basophils produce IL-4 and accumulate in tissues after infection with a Th2-inducing parasite. J. Exp. Med. 2004, 200, 507–517.
- Imran, M.; Laddha, N.C.; Dwivedi, M.; Mansuri, M.S.; Singh, J.; Rani, R.; Gokhale, R.S.; Sharma, V.K.; Marfatia, Y.S.; Begum, R. Interleukin-4 genetic variants correlate with its transcript and protein levels in patients with vitiligo. Br. J. Dermatol. 2012, 167, 314–323.
- Salgame, P.; Abrams, J.S.; Clayberger, C.; Goldstein, H.; Convit, J.; Modlin, R.L.; Bloom, B.R. Differing lymphokine profiles of functional subsets of human CD4 and CD8 T cell clones. Science 1991, 254, 279–282.
- Basak, P.Y.; Adiloglu, A.K.; Ceyhan, A.M.; Tas, T.; Akkaya, V.B. The role of helper and regulatory T cells in the pathogenesis of vitiligo. J. Am. Acad. Dermatol. 2009, 60, 256–260.
- Nouri-Koupaee, A.; Mansouri, P.; Jahanbini, H.; Sanati, M.H.; Jadali, Z. Differential expression of mRNA for T-bet and GATA-3 transcription factors in peripheral blood mononuclear cells of patients with vitiligo. Clin. Exp. Dermatol. 2015, 40, 735–740.
- Rael, E.L.; Lockey, R.F. Interleukin-13 signaling and its role in asthma. World Allergy Organ J. 2011, 4, 54–64.
- Nishimura, Y.; Nitto, T.; Inoue, T.; Node, K. IL-13 attenuates vascular tube formation via JAK2-STAT6 pathway. Circ. J. 2008, 72, 469–475.
- Renauld, J.C. New insights into the role of cytokines in asthma. J. Clin. Pathol. 2001, 54, 577–589.
- Kim, K.; Han, J.; Lee, T.R.; Shin, D.W.; Chang, H.; Cho, A.R.; Choi, S.J.; Jo, S.J.; Kwon, O. Comparative Analysis of Human Epidermal and Peripheral Blood γδ T Cell Cytokine Profiles. Ann. Dermatol. 2014, 26, 308–313.
- Han, J.; Lee, E.; Kim, E.; Yeom, M.H.; Kwon, O.; Yoon, T.H.; Lee, T.R.; Kim, K. Role of epidermal γδ T-cell-derived interleukin 13 in the skin-whitening effect of Ginsenoside F1. Exp. Dermatol. 2014, 23, 860–862.
- Speeckaert, R.; Lambert, J.; Grine, L.; Van Gele, M.; De Schepper, S.; van Geel, N. The many faces of interleukin-17 in inflammatory skin diseases. Br. J. Dermatol. 2016, 175, 892–901.
- Volpe, E.; Servant, N.; Zollinger, R.; Bogiatzi, S.I.; Hupé, P.; Barillot, E.; Soumelis, V. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat. Immunol. 2008, 9, 650–657.
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279.
- Meephansan, J.; Komine, M.; Tsuda, H.; Tominaga, S.; Ohtsuki, M. Ultraviolet B irradiation induces the expression of IL-33 mRNA and protein in normal human epidermal keratinocytes. J. Dermatol. Sci. 2012, 65, 72–74.
- Zhou, J.; Song, J.; Ping, F.; Shang, J. Enhancement of the p38 MAPK and PKA signaling pathways is associated with the pro-melanogenic activity of Interleukin 33 in primary melanocytes. J. Dermatol. Sci. 2014, 73, 110–116.
- Zhou, J.; Shang, J.; Song, J.; Ping, F. Interleukin-18 augments growth ability of primary human melanocytes by PTEN inactivation through the AKT/NF-κB pathway. Int. J. Biochem. Cell Biol. 2013, 45, 308–316.
- Chen, N.; Hu, Y.; Li, W.H.; Eisinger, M.; Seiberg, M.; Lin, C.B. The role of keratinocyte growth factor in melanogenesis: A possible mechanism for the initiation of solar lentigines. Exp. Dermatol. 2010, 19, 865–872.
- Kholmanskikh, O.; van Baren, N.; Brasseur, F.; Ottaviani, S.; Vanacker, J.; Arts, N.; van der Bruggen, P.; Coulie, P.; De Plaen, E. Interleukins 1alpha and 1beta secreted by some melanoma cell lines strongly reduce expression of MITF-M and melanocyte differentiation antigens. Int. J. Cancer 2010, 127, 1625–1636.
- Scott, G.; Leopardi, S.; Printup, S.; Malhi, N.; Seiberg, M.; Lapoint, R. Proteinase-activated receptor-2 stimulates prostaglandin production in keratinocytes: Analysis of prostaglandin receptors on human melanocytes and effects of PGE2 and PGF2alpha on melanocyte dendricity. J. Investig. Dermatol. 2004, 122, 1214–1224.
- Natarajan, V.T.; Ganju, P.; Singh, A.; Vijayan, V.; Kirty, K.; Yadav, S.; Puntambekar, S.; Bajaj, S.; Dani, P.P.; Kar, H.K.; et al. IFN-γ signaling maintains skin pigmentation homeostasis through regulation of melanosome maturation. Proc. Natl. Acad. Sci. USA 2014, 111, 2301–2306.
- Zhou, J.; Ling, J.; Wang, Y.; Shang, J.; Ping, F. Cross-talk between interferon-gamma and interleukin-18 in melanogenesis. J. Photochem. Photobiol. B 2016, 163, 133–143.
- Videira, I.F.; Moura, D.F.; Magina, S. Mechanisms regulating melanogenesis. An. Bras. Dermatol. 2013, 88, 76–83.
- Arend, W.P.; Palmer, G.; Gabay, C. IL-1, IL-18, and IL-33 families of cytokines. Immunol. Rev. 2008, 223, 20–38.
- Byrne, S.N.; Beaugie, C.; O’Sullivan, C.; Leighton, S.; Halliday, G.M. The immune-modulating cytokine and endogenous Alarmin interleukin-33 is upregulated in skin exposed to inflammatory UVB radiation. Am. J. Pathol. 2011, 179, 211–222.
- Ali, S.; Huber, M.; Kollewe, C.; Bischoff, S.C.; Falk, W.; Martin, M.U. IL-1 receptor accessory protein is essential for IL-33-induced activation of T lymphocytes and mast cells. Proc. Natl. Acad. Sci. USA 2007, 104, 18660–18665.
- Allakhverdi, Z.; Smith, D.E.; Comeau, M.R.; Delespesse, G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J. Immunol. 2007, 179, 2051–2054.
- Moulin, D.; Donzé, O.; Talabot-Ayer, D.; Mézin, F.; Palmer, G.; Gabay, C. Interleukin (IL)-33 induces the release of pro-inflammatory mediators by mast cells. Cytokine 2007, 40, 216–225.
- Theoharides, T.C.; Zhang, B.; Kempuraj, D.; Tagen, M.; Vasiadi, M.; Angelidou, A.; Alysandratos, K.D.; Kalogeromitros, D.; Asadi, S.; Stavrianeas, N.; et al. IL-33 augments substance P-induced VEGF secretion from human mast cells and is increased in psoriatic skin. Proc. Natl. Acad. Sci. USA 2010, 107, 4448–4453.
- Pushparaj, P.N.; Tay, H.K.; H’Ng, S.C.; Pitman, N.; Xu, D.; McKenzie, A.; Liew, F.Y.; Melendez, A.J. The cytokine interleukin-33 mediates anaphylactic shock. Proc. Natl. Acad. Sci. USA 2009, 106, 9773–9778.
- Kurowska-Stolarska, M.; Stolarski, B.; Kewin, P.; Murphy, G.; Corrigan, C.J.; Ying, S.; Pitman, N.; Mirchandani, A.; Rana, B.; van Rooijen, N.; et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J. Immunol. 2009, 183, 6469–6477.
- Ohno, T.; Oboki, K.; Kajiwara, N.; Morii, E.; Aozasa, K.; Flavell, R.A.; Okumura, K.; Saito, H.; Nakae, S. Caspase-1, caspase-8, and calpain are dispensable for IL-33 release by macrophages. J. Immunol. 2009, 183, 7890–7897.
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490.
- Hueber, A.J.; Alves-Filho, J.C.; Asquith, D.L.; Michels, C.; Millar, N.L.; Reilly, J.H.; Graham, G.J.; Liew, F.Y.; Miller, A.M.; McInnes, I.B. IL-33 induces skin inflammation with mast cell and neutrophil activation. Eur. J. Immunol. 2011, 41, 2229–2237.
- Suzukawa, M.; Iikura, M.; Koketsu, R.; Nagase, H.; Tamura, C.; Komiya, A.; Nakae, S.; Matsushima, K.; Ohta, K.; Yamamoto, K.; et al. An.IL-1 cytokine member, IL-33, induces human basophil activation via its ST2 receptor. J. Immunol. 2008, 181, 5981–5989.
- Rank, M.A.; Kobayashi, T.; Kozaki, H.; Bartemes, K.R.; Squillace, D.L.; Kita, H. IL-33-activated dendritic cells induce an atypical TH2-type response. J. Allergy Clin. Immunol. 2009, 123, 1047–1054.
- Hsu, C.L.; Chhiba, K.D.; Krier-Burris, R.; Hosakoppal, S.; Berdnikovs, S.; Miller, M.L.; Bryce, P.J. Allergic inflammation is initiated by IL-33-dependent crosstalk between mast cells and basophils. PLoS ONE 2020, 15, e0226701.
- Cevikbas, F.; Steinhoff, M. IL-33: A novel danger signal system in atopic dermatitis. J. Investig. Dermatol. 2012, 132, 1326–1329.
- Dinarello, C.A. Interleukin-1. Rev. Infect. Dis. 1984, 6, 51–95.
- Kampschmidt, R.F. The numerous postulated biological manifestations of interleukin-1. J. Leukoc. Biol. 1984, 36, 341–355.
- Kupper, T.S. Immune and inflammatory processes in cutaneous tissues. Mechanisms and speculations. J. Clin. Investig. 1990, 86, 1783–1789.
- Auron, P.E.; Webb, A.C.; Rosenwasser, L.J.; Mucci, S.F.; Rich, A.; Wolff, S.M.; Dinarello, C.A. Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc. Natl. Acad. Sci. USA 1984, 81, 7907–7911.
- Sims, J.E.; March, C.J.; Cosman, D.; Widmer, M.B.; MacDonald, H.R.; McMahan, C.J.; Grubin, C.E.; Wignall, J.M.; Jackson, J.L.; Call, S.M.; et al. cDNA expression cloning of the IL-1 receptor, a member of the immunoglobulin superfamily. Science 1988, 241, 585–589.
- March, C.J.; Mosley, B.; Larsen, A.; Cerretti, D.P.; Braedt, G.; Price, V.; Gillis, S.; Henney, C.S.; Kronheim, S.R.; Grabstein, K.; et al. Cloning, sequence and expression of two distinct human interleukin-1 complementary DNAs. Nature 1985, 315, 641–647.
- Groves, R.W.; Sherman, L.; Mizutani, H.; Dower, S.K.; Kupper, T.S. Detection of interleukin-1 receptors in human epidermis. Induction of the type II receptor after organ culture and in psoriasis. Am. J. Pathol. 1994, 145, 1048–1056.
- Martin, M.U.; Wesche, H. Summary and comparison of the signaling mechanisms of the Toll/interleukin-1 receptor family. Biochim. Biophys. Acta 2002, 1592, 265–280.
- Tang, A.; Gilchrest, B.A. Regulation of keratinocyte growth factor gene expression in human skin fibroblasts. J. Dermatol. Sci. 1996, 11, 41–50.
- Groves, R.W.; Ross, E.; Barker, J.N.; Ross, J.S.; Camp, R.D.; MacDonald, D.M. Effect of in vivo interleukin-1 on adhesion molecule expression in normal human skin. J. Investig. Dermatol. 1992, 98, 384–387.
- Kondo, S.; Sauder, D.N.; Kono, T.; Galley, K.A.; McKenzie, R.C. Differential modulation of interleukin-1 alpha (IL-1 alpha) and interleukin-1 beta (IL-1 beta) in human epidermal keratinocytes by UVB. Exp. Dermatol. 1994, 3, 29–39.
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556.
- Pentland, A.P.; Mahoney, M.; Jacobs, S.C.; Holtzman, M.J. Enhanced prostaglandin synthesis after ultraviolet injury is mediated by endogenous histamine stimulation. A mechanism for irradiation erythema. J. Clin. Investig. 1990, 86, 566–574.
- Pentland, A.P.; Mahoney, M.G. Keratinocyte prostaglandin synthesis is enhanced by IL-1. J. Investig. Dermatol. 1990, 94, 43–46.
- Fosslien, E. Molecular pathology of cyclooxygenase-2 in neoplasia. Ann. Clin. Lab. Sci. 2000, 30, 3–21.
- Pentland, A.P.; Needleman, P. Modulation of keratinocyte proliferation in vitro by endogenous prostaglandin synthesis. J. Clin. Investig. 1986, 77, 246–251.
- Scott, G.; Jacobs, S.; Leopardi, S.; Anthony, F.A.; Learn, D.; Malaviya, R.; Pentland, A. Effects of PGF2alpha on human melanocytes and regulation of the FP receptor by ultraviolet radiation. Exp. Cell Res. 2005, 304, 407–416.
- Holtzman, M.J.; Zhang, V.; Hussain, H.; Roswit, W.T.; Wilson, J.D. Prostaglandin H synthase and lipoxygenase gene families in the epithelial cell barrier. Ann. N. Y. Acad. Sci. 1994, 744, 58–77.
- Bach, E.A.; Aguet, M.; Schreiber, R.D. The IFN gamma receptor: A paradigm for cytokine receptor signaling. Annu. Rev. Immunol. 1997, 15, 563–591.
- Carnaud, C.; Lee, D.; Donnars, O.; Park, S.H.; Beavis, A.; Koezuka, Y.; Bendelac, A. Cutting edge: Cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J. Immunol. 1999, 163, 4647–4650.
- Frucht, D.M.; Fukao, T.; Bogdan, C.; Schindler, H.; O’Shea, J.J.; Koyasu, S. IFN-gamma production by antigen-presenting cells: Mechanisms emerge. Trends Immunol. 2001, 22, 556–560.
- Flaishon, L.; Hershkoviz, R.; Lantner, F.; Lider, O.; Alon, R.; Levo, Y.; Flavell, R.A.; Shachar, I. Autocrine secretion of interferon gamma negatively regulates homing of immature B cells. J. Exp. Med. 2000, 192, 1381–1388.
- Nishimura, E.K.; Suzuki, M.; Igras, V.; Du, J.; Lonning, S.; Miyachi, Y.; Roes, J.; Beermann, F.; Fisher, D.E. Key roles for transforming growth factor beta in melanocyte stem cell maintenance. Cell Stem Cell 2010, 6, 130–140.
- Abdel-Malek, Z.; Scott, M.C.; Suzuki, I.; Tada, A.; Im, S.; Lamoreux, L.; Ito, S.; Barsh, G.; Hearing, V.J. The melanocortin-1 receptor is a key regulator of human cutaneous pigmentation. Pigment. Cell Res. 2000, 13 (Suppl. 8) (Suppl. 8), 156–162.
- Harris, J.E.; Harris, T.H.; Weninger, W.; Wherry, E.J.; Hunter, C.A.; Turka, L.A. A mouse model of vitiligo with focused epidermal depigmentation requires IFN-γ for autoreactive CD8+ T-cell accumulation in the skin. J. Investig. Dermatol. 2012, 132, 1869–1876.
- Gregg, R.K.; Nichols, L.; Chen, Y.; Lu, B.; Engelhard, V.H. Mechanisms of spatial and temporal development of autoimmune vitiligo in tyrosinase-specific TCR transgenic mice. J. Immunol. 2010, 184, 1909–1917.
- Yang, L.; Wei, Y.; Sun, Y.; Shi, W.; Yang, J.; Zhu, L.; Li, M. Interferon-gamma Inhibits Melanogenesis and Induces Apoptosis in Melanocytes: A Pivotal Role of CD8+ Cytotoxic T Lymphocytes in Vitiligo. Acta Derm. Venereol. 2015, 95, 664–670.
- Kristensen, M.; Chu, C.Q.; Eedy, D.J.; Feldmann, M.; Brennan, F.M.; Breathnach, S.M. Localization of tumour necrosis factor-alpha (TNF-alpha) and its receptors in normal and psoriatic skin: Epidermal cells express the 55-kD but not the 75-kD TNF receptor. Clin. Exp. Immunol. 1993, 94, 354–362.
- Meephansan, J.; Tsuda, H.; Komine, M.; Tominaga, S.; Ohtsuki, M. Regulation of IL-33 expression by IFN-γ and tumor necrosis factor-α in normal human epidermal keratinocytes. J. Investig. Dermatol. 2012, 132, 2593–2600.
- Meephansan, J.; Komine, M.; Tsuda, H.; Karakawa, M.; Tominaga, S.; Ohtsuki, M. Expression of IL-33 in the epidermis: The mechanism of induction by IL-17. J. Dermatol. Sci. 2013, 71, 107–114.
- Park, K.C.; Kim, D.S.; Kim, H.J.; Seo, K.I.; Kim, K.H.; Chung, J.H.; Eun, H.C.; Jung, H.C. Growth related secretion and production of GM-CSF by epithelial cell line. J. Dermatol. Sci. 2001, 25, 53–58.
- Wu, X.G.; Hong, W.S.; Xu, A. GM-CSF: A possible prognostic serum biomarker of vitiligo patients’ considered for transplantation treatment with cultured autologous melanocytes: A pilot study. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1409–1411.