Neurodegenerative diseases (NDs) including Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and Huntington’s disease are incurable and affect millions of people worldwide. The development of treatments for this unmet clinical need is a major global research challenge. Computer-aided drug design (CADD) methods minimize the huge number of ligands that could be screened in biological assays, reducing the cost, time, and effort required to develop new drugs.
- neurodegeneration
- drug discovery
- CADD
- dementia
- brain diseases
- CNS disorders
- Alzheimer’s disease
- Parkinson’s disease
- amyotrophic lateral sclerosis
- Huntington’s disease
1. Introduction
Neurodegenerative diseases (NDs) are incurable and debilitating conditions that result in progressive degeneration and/or death of nerve cells in the central nervous system (CNS) [1][2][3]. Dementia rates are alarmingly on the rise worldwide. There are over 50 million people worldwide living with dementia in 2020, with nearly 60% living in low- and middle-income countries [4]. This number will almost double every 20 years, reaching 82 million in 2030 and 152 million in 2050 [4]. The number of people with dementia in the UK is predicted to be around 1.14 million by 2025 and 2.1 million by 2051, an increase of 40% over the next 5 years and 157% over the next 31 years [5].
The UK Prime Minister’s Challenge on Dementia was launched in 2015 to identify strategies to tackle dementia by 2025 [6]. Current therapies for NDs treat symptoms, not the underlying pathological changes. There is a clear and unmet clinical need to develop new therapies based on understanding the molecular pathologies. One of the most promising approaches is to develop novel therapeutics using computer-aided drug design (CADD) [7][8].
2. Computer-Aided Drug Design
“Computer-aided drug design” (CADD) refers to the application of computational modelling approaches to drug discovery. Drug discovery is an expensive and time-consuming process with the average approved drug requiring 10 to 15 years to develop with an estimated cost of 0.8–2 billion USD [9]. Many licensed drugs, such as captopril, dorzolamide, oseltamivir, aliskiren, and nolatrexed, were all optimized using CADD [10], and a large number of publications describe the successful design and discovery of leads/drugs using CADD [11][12][13]. The major steps involved in CADD are summarized in Figure 1A and discussed in the following sections. The main goal of CADD is to reduce these timescales and costs without affecting quality (Figure 1B) [14]. Importantly, CADD can be used in most stages of drug development: from target identification to target validation, from lead discovery to optimization, and in preclinical studies. It is therefore estimated that CADD could reduce the cost of drug development by up to 50% [15][16].
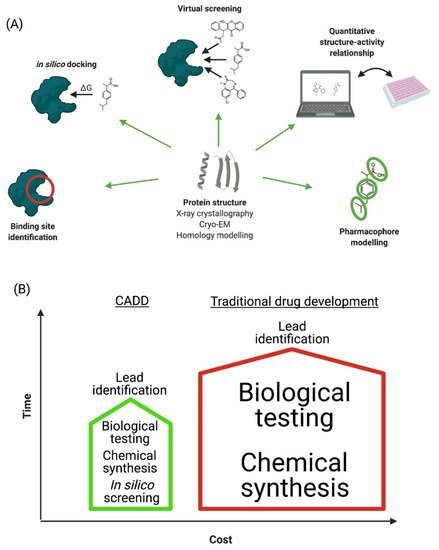
Figure 1. (A) Schematic representation of CADD process. (B) Comparison of traditional and computer-aided drug development in terms of time and cost investments.
2.1. Drug Target Selection
Drug target selection is the first step of structure-based drug design. This involves identifying and determining the structures of the relevant proteins [17]. Understanding and characterization of the molecular biology of the targeted disease are therefore necessary before the initiation of any active compound search process.
2.2. Determination of the Protein Structure
An in-depth understanding of biological processes is still often hampered by a lack of detailed knowledge of protein structures [18]. The determination of the structure of the target protein is a prerequisite for CADD [19]. Structural elucidation of the target protein can be performed by experimental tools including, but not limited to, nuclear magnetic resonance (NMR) spectroscopy, Cryo-EM, and X-ray crystallography [20][21].
2.3. Homology Modelling
Despite the current revolution in structural studies, in particular the recent developments in cryo-EM, the detailed structures of a large number of proteins, and especially membrane proteins (which are over-represented amongst drug targets), have not been determined [18][22]. Homology modelling is an approach to estimate the structure of a target protein based on structural data from proteins with sequence homology to the target [23].
For instance, a homology model of human catechol-O-methyltransferase (COMT) was constructed utilizing the X-ray crystal structure of rat COMT to design anti-PD drugs by performing ligand docking, resulting in the discovery of nine putative inhibitors. Another example involves a cysteine protease from Xanthomonas campestris (an aerobic, Gram-negative rod-shaped bacterium known to cause black rot in crucifers by darkening the vascular tissues). The active site of this enzyme is homologous to human cathepsin B enzyme (hCB), the activity of which contributes to the reduction of the amyloid peptide by proteolytic cleavage of Aβ1-42, offering a protective role against AD [24].
2.4. Identification of Binding Sites
When the three-dimensional structure of the target protein is determined, the next step is the identification of potential binding sites for small molecules. This process can be conducted using various algorithms for computing and identifying binding pockets [25][26][27].
2.5. Molecular Dynamics Simulation
Molecular dynamics (MD) simulations are a theoretical tool to discover the configurations and dynamic behaviours of molecules, providing atomic-level insight into drug mechanisms of action [13]. MD may also help to reveal the aggregation pathway of neurotoxic protein aggregates and thus aid in the design of new inhibitors [28].
2.6. Molecular Docking Studies
Molecular docking is a computational procedure that predicts the lowest energy binding conformations of one molecule to a second (usually a small drug-like molecule to a protein). Accordingly, molecular docking procedures, along with their different scoring systems, are frequently utilized to predict the binding modes and affinities between chemical compounds and drug binding sites on biological macromolecules [29][30].
2.7. Virtual Screening
Virtual screening (VS) is the process of screening small molecule libraries in silico to identify chemical structures that may bind to a drug target [31][32][33].
2.8. Quantitative Structure—Activity Relationship Study
Quantitative structure—activity relationship (QSAR) methods are conducted to correlate a biological response (e.g., enzyme activity, cell viability, etc.) to the chemical properties of a set of molecules [34][35][36].
2.9. Pharmacophore Modelling
Pharmacophore modelling deals with finding the optimal shapes and charge distributions for binding of a small molecule to a biological macromolecule. Pharmacophore modelling is commonly implemented to rapidly specify potential lead compounds [37][38].
3. A Roadmap for Implementing CADD in ND Drug Design
Even with the number of successful implementations of CADD in modern drug discovery, it has its limitations. Molecules designed in silico utilizing computational and theoretical chemistry sometimes do not work in real biological systems [39][40]. In general, poor pharmacokinetics and/or pharmacodynamics result in only 40% of drug candidates passing phase I clinical trials [41]. Moreover, each computational technique depends on pre-determined algorithms that have their own limitations. CADD results must be validated in real biological systems, as many molecules that appear to bind in silico do not show the desired activity in vitro. Another limitation of CADD is that all tools for designing and discovery of new drugs are based on algorithms that, by necessity, simplify the underlying physics and chemistry and, therefore, have a variety of limitations that necessitate the continuous updating of these algorithms to enhance the accuracy and thus the provision of new drugs [42][43][44][45][46][47][48]. Furthermore, the shortage of experimental data regarding predicted absorption, distribution, metabolism, excretion, and toxicity results has led to several published failures [49][50][51][52][53].
To overcome the limitations and improve the accuracy of CADD it is necessary to update and develop software and associated algorithms, validate with experimental data, use reliable databases (e.g., PDB), and use algorithms that give docking scores that accurately predict in vitro binding with comprehensive and fully retrospective coverage of the published literature [54][55][56]. For example, by September 2020, the Cambridge Structural Dataset (CSD) acquired more than 1.8 million entries, which may help with future developments in small molecule structural modelling [57]. Consequently, the above-mentioned tools could help with future design of pharmacophores that possess the desired biological activity [58][59][60].
One of the main reasons for implementing in silico drug design is to predict the ligand–target binding in terms of binding site and binding strength. To predict potential ligands to treat NDs, novel target proteins must be identified and studied, and the resulting docking studies should be validated in vitro and eventually in the clinic [61][62][63].
In the meantime, there is no effective treatment to cure NDs, although many treatments are available that offer minor improvement of symptoms [2]. The development of effective treatments is further hindered by the BBB that excludes many molecules from the CNS parenchyma [64][65][66]. Accordingly, clinical effectiveness of a potential drug is not guaranteed even with positive data in silico, in vitro, and in vivo [67][68][69][70].
New experimental approaches including genome-wide association studies (GWAS) [68][71][72], CRISPR-Cas9 technology [73][74][75], high throughput screening (HTS) [76], organ-on-chip technologies [77][78], functional MRI (fMRI) techniques [79][80], and positron emission tomography (PET) [81] may lead to new drug targets for NDs, which can feed into future CADD projects.
Being incurable, the NDs are major challenges to healthcare providers and research scientists. The accelerating increase in the numbers of affected people adds more impetus to tackle NDs. Developing a better understanding of NDs and the underlying molecular pathophysiology will provide more opportunities to develop novel treatments in the near future. This may be achieved with the incorporation of computational tools. CADD can have a major impact on drug discovery by saving both time and money and reducing the risk of following up with the development of non-viable leads.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22094688
References
- Maciejczyk, M.; Zalewska, A. Salivary redox biomarkers in selected neurodegenerative diseases. J. Clin. Med. 2020, 9, 497.
- Rabanel, J.-M.; Perrotte, M.; Ramassamy, C. Nanotechnology at the Rescue of Neurodegenerative Diseases: Tools for Early Diagnostic. In Nanobiotechnology in Neurodegenerative Diseases; Springer: Berlin/Heidelberg, Germany, 2019; pp. 19–48.
- Sehgal, S.A.; Hammad, M.A.; Tahir, R.A.; Akram, H.N.; Ahmad, F. Current Therapeutic Molecules and Targets in Neurodegenerative Diseases Based on in silico Drug Design. Curr. Neuropharmacol. 2018, 16, 649–663.
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446.
- Prince, M.; Knapp, M.; Guerchet, M.; McCrone, P.; Prina, M.; Comas-Herrera, A.; Wittenberg, R.; Adelaja, B.; Hu, B.; King, D. Dementia UK: Update; Alzheimers Society: Belfast, UK, 2014; pp. 1–136.
- Prime Minister’s Challenge on Dementia. Available online: (accessed on 14 April 2020).
- Shukla, R.; Singh, T.R. Virtual screening, pharmacokinetics, molecular dynamics and binding free energy analysis for small natural molecules against cyclin-dependent kinase 5 for Alzheimer’s disease. J. Biomol. Struct. Dyn. 2020, 38, 248–262.
- Mouchlis, V.D.; Melagraki, G.; Zacharia, L.C.; Afantitis, A. Computer-Aided Drug Design of β-Secretase, γ-Secretase and Anti-Tau Inhibitors for the Discovery of Novel Alzheimer’s Therapeutics. Int. J. Mol. Sci. 2020, 21, 703.
- Am Ende, D.J.; Am Ende, M.T. Chemical engineering in the pharmaceutical industry: An introduction. Chem. Eng. Pharm. Ind. Drug Prod. Des. Dev. Modeling 2019, 1–17.
- Talele, T.T.; Khedkar, S.A.; Rigby, A.C. Successful applications of computer aided drug discovery: Moving drugs from concept to the clinic. Curr. Top. Med. Chem. 2010, 10, 127–141.
- Kaushik, A.C.; Kumar, A.; Bharadwaj, S.; Chaudhary, R.; Sahi, S. Structure-Based Approach for In-silico Drug Designing. In Bioinformatics Techniques for Drug Discovery; Springer: Berlin/Heidelberg, Germany, 2018; pp. 21–25.
- Hamad, O.; Amran, S.; Sabbah, A. Drug Discovery-Yesterday and Tomorrow: The Common Approaches in Drug Design and Cancer. Cell Cell. Life Sci. J. 2018, 3, 000119.
- Lu, W.; Zhang, R.; Jiang, H.; Zhang, H.; Luo, C. Computer-Aided Drug Design in Epigenetics. Front. Chem. 2018, 6.
- Kapetanovic, I. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem. Biol. Interact. 2008, 171, 165–176.
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharmacal. Res. 2015, 38, 1686–1701.
- Xiang, M.; Cao, Y.; Fan, W.; Chen, L.; Mo, Y. Computer-aided drug design: Lead discovery and optimization. Comb. Chem. High Throughput Screen. 2012, 15, 328–337.
- Anderson, A.; Blaney, J.; Blundell, T.; Clark, D.; Davis, A.M.; Ealick, S.; Kim, S.-H.; McCammon, J.A.; Verdonk, M.; Wijnand, M. Computational and Structural Approaches to Drug Discovery: Ligand-Protein Interactions; Royal Society of Chemistry: London, UK, 2007.
- Hauri, S.; Khakzad, H.; Happonen, L.; Teleman, J.; Malmström, J.; Malmström, L. Rapid determination of quaternary protein structures in complex biological samples. Nat. Commun. 2019, 10, 1–10.
- Baig, M.H.; Ahmad, K.; Rabbani, G.; Danishuddin, M.; Choi, I. Computer aided drug design and its application to the development of potential drugs for neurodegenerative disorders. Curr. Neuropharmacol. 2018, 16, 740–748.
- Makhouri, F.R.; Ghasemi, J.B. In Silico studies in drug research against neurodegenerative diseases. Curr. Neuropharmacol. 2018, 16, 664–725.
- Fawzi, M.M.; Abdallah, H.H.; Suroowan, S.; Jugreet, S.; Zhang, Y.; Hu, X. In Silico Exploration of Bioactive Phytochemicals Against Neurodegenerative Diseases via Inhibition of Cholinesterases. Curr. Pharm. Des. 2020, 26, 4151–4162.
- Schmiedel, J.M.; Lehner, B. Determining protein structures using deep mutagenesis. Nat. Genet. 2019, 51, 1177.
- Kumar, J.; Ranjan, T.; Kumar, R.R.; Ansar, M.; Rajani, K.; Kumar, M.; Kumar, V.; Kumar, A. In silico Characterization and Homology Modelling of Potato Leaf Roll Virus (PLRV) Coat Protein. Curr. J. Appl. Sci. Technol. 2019, 1–8.
- Morales-Navarro, S.; Prent-Peñaloza, L.; Rodríguez Núñez, Y.A.; Sánchez-Aros, L.; Forero-Doria, O.; González, W.; Campilllo, N.E.; Reyes-Parada, M.; Martínez, A.; Ramírez, D. Theoretical and Experimental Approaches AiMed. at Drug Design Targeting Neurodegenerative Diseases. Processes 2019, 7, 940.
- Wang, B.; Dai, P.; Ding, D.; Del Rosario, A.; Grant, R.A.; Pentelute, B.L.; Laub, M.T. Affinity-based capture and identification of protein effectors of the growth regulator ppGpp. Nat. Chem. Biol. 2019, 15, 141–150.
- Chan, H.S.; Li, Y.; Dahoun, T.; Vogel, H.; Yuan, S. New binding sites, new opportunities for GPCR drug discovery. Trends Biochem. Sci. 2019, 44, 312–330.
- Zhang, Y.; Qiao, S.; Ji, S.; Han, N.; Liu, D.; Zhou, J. Identification of DNA–protein binding sites by bootstrap multiple convolutional neural networks on sequence information. Eng. Appl. Artif. Intell. 2019, 79, 58–66.
- Ye, W.; Wang, W.; Jiang, C.; Yu, Q.; Chen, H. Molecular dynamics simulations of amyloid fibrils: An in silico approach. Acta Biochim. Biophys. Sin. 2013, 45, 503–508.
- Makarasen, A.; Kuno, M.; Patnin, S.; Reukngam, N.; Khlaychan, P.; Deeyohe, S.; Intachote, P.; Saimanee, B.; Sengsai, S.; Boonsri, P. Molecular Docking Studies and Synthesis of Amino-oxy-diarylquinoline Derivatives as Potent Non-nucleoside HIV-1 Reverse Transcriptase Inhibitors. Drug Res. 2019, 69, 671–682.
- Vilar, S.; Sobarzo-Sánchez, E.; Uriarte, E. In Silico Prediction of P-glycoprotein Binding: Insights from Molecular Docking Studies. Curr. Med. Chem. 2019, 26, 1746–1760.
- Nunes, R.R.; Fonseca, A.L.D.; Pinto, A.C.D.S.; Maia, E.H.B.; Silva, A.M.D.; Varotti, F.D.P.; Taranto, A.G. Brazilian malaria molecular targets (BraMMT): Selected receptors for virtual high-throughput screening experiments. Memórias Do Inst. Oswaldo Cruz 2019, 114, e180465.
- Zerroug, A.; Belaidi, S.; BenBrahim, I.; Sinha, L.; Chtita, S. Virtual screening in drug-likeness and structure/activity relationship of pyridazine derivatives as Anti-Alzheimer drugs. J. King Saud Univ. Sci. 2019, 31, 595–601.
- Vieira, T.; Magalhaes, R.; Sousa, S. Tailoring specialized scoring functions for more efficient virtual screening. Frontiers 2019, 2, 1–4.
- Ray, R. Understanding the Structural Importance of the Non-Binding and Binding Parts of Bedaquiline and Its Analogues with ATP Synthase Subunit C Using Molecular Docking, Molecular Dynamics Simulation and 3D-QSAR Techniques. In Proceedings of the International Conference on Drug Discovery (ICDD) 2020, Hyderabad, India, 19 February 2020.
- Kotzabasaki, M.I.; Sotiropoulos, I.; Sarimveis, H. QSAR modeling of the toxicity classification of superparamagnetic iron oxide nanoparticles (SPIONs) in stem-cell monitoring applications: An integrated study from data curation to model development. RSC Adv. 2020, 10, 5385–5391.
- Gbeddy, G.; Egodawatta, P.; Goonetilleke, A.; Ayoko, G.; Chen, L. Application of quantitative structure-activity relationship (QSAR) model in comprehensive human health risk assessment of PAHs, and alkyl-, nitro-, carbonyl-, and hydroxyl-PAHs laden in urban road dust. J. Hazard. Mater. 2020, 383, 121154.
- Du, M.; Qiu, Y.; Li, Q.; Li, Y. Efficacy coefficient method assisted quadruple-activities 3D-QSAR pharmacophore model for application in environmentally friendly PAE molecular modification. Environ. Sci. Pollut. Res. Int. 2020, 27, 24103–24114.
- Fan, F.; Warshaviak, D.T.; Hamadeh, H.K.; Dunn, R.T. The integration of pharmacophore-based 3D QSAR modeling and virtual screening in safety profiling: A case study to identify antagonistic activities against adenosine receptor, A2A, using 1,897 known drugs. PLoS ONE 2019, 14, e0204378.
- Schneider, G. Virtual screening: An endless staircase? Nat. Rev. Drug Discov. 2010, 9, 273–276.
- Verkhivker, G.M.; Bouzida, D.; Gehlhaar, D.K.; Rejto, P.A.; Arthurs, S.; Colson, A.B.; Freer, S.T.; Larson, V.; Luty, B.A.; Marrone, T. Deciphering common failures in molecular docking of ligand-protein complexes. J. Comput. Aided Mol. Des. 2000, 14, 731–751.
- Josephs, D.; Spicer, J.; O’Doherty, M. Molecular imaging in clinical trials. Target. Oncol. 2009, 4, 151–168.
- Cheatham, T.E., III; Young, M.A. Molecular dynamics simulation of nucleic acids: Successes, limitations, and promise. Biopolym. Orig. Res. Biomol. 2000, 56, 232–256.
- Klebe, G. Virtual ligand screening: Strategies, perspectives and limitations. Drug Discov. Today 2006, 11, 580–594.
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421.
- Korb, O.; Olsson, T.S.; Bowden, S.J.; Hall, R.J.; Verdonk, M.L.; Liebeschuetz, J.W.; Cole, J.C. Potential and limitations of ensemble docking. J. Chem. Inf. Modeling 2012, 52, 1262–1274.
- MacDonald, D.; Breton, R.; Sutcliffe, R.; Walker, J. Uses and limitations of quantitative structure-activity relationships (QSARs) to categorize substances on the Canadian Domestic Substance List as persistent and/or bioaccumulative, and inherently toxic to non-human organisms. Sar Qsar Environ. Res. 2002, 13, 43–55.
- Papakonstantinou, E.; Megalooikonomou, V.; Vlachakis, D. Dark Suite: A comprehensive toolbox for computer-aided drug design. Embnet. J. 2020, 25, e928.
- Berdigaliyev, N.; Aljofan, M. An overview of drug discovery and development. Future Med. Chem. 2020, 12, 939–947.
- Shi, J.; Zha, W. Predicting Human Pharmacokinetics: Physiologically Based Pharmacokinetic Modeling and In Silico ADME Prediction in Early Drug Discovery. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 135–137.
- Van De Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204.
- Faller, B.; Winiwarter, S.; Chang, G.; Desai, P.; Menzel, C.; Rieko, A.; Keefer, C.; Broccatelli, F. Prediction of fraction unbound in microsomal and hepatocyte incubations–a comparison of methods across industry data sets (by the IQ in silico ADME working group). Mol. Pharm. 2019, 16, 4077–4085.
- Blomme, E.A.; Will, Y. Toxicology strategies for drug discovery: Present and future. Chem. Res. Toxicol. 2016, 29, 473–504.
- Pruss, R.M. Phenotypic screening strategies for neurodegenerative diseases: A pathway to discover novel drug candidates and potential disease targets or mechanisms. CNS Neurol. Disord. Drug Targets Former. Curr. Drug Targets CNS Neurol. Disord. 2010, 9, 693–700.
- Ren, J.; Wen, L.; Gao, X.; Jin, C.; Xue, Y.; Yao, X. CSS-Palm 2.0: An updated software for palmitoylation sites prediction. Protein Eng. Des. Sel. 2008, 21, 639–644.
- Nikzad, H.; Karimian, M.; Sareban, K.; Khoshsokhan, M.; Colagar, A.H. MTHFR-Ala222Val and male infertility: A study in Iranian men, an updated meta-analysis and an in silico-analysis. Reprod. Biomed. Online 2015, 31, 668–680.
- Glykos, N.M. Software news and updates carma: A molecular dynamics analysis program. J. Comput. Chem. 2006, 27, 1765–1768.
- CCDC. What’s New. Available online: (accessed on 14 April 2021).
- Zuo, Z.; MacMillan, D.W. Decarboxylative arylation of α-amino acids via photoredox catalysis: A one-step conversion of biomass to drug pharmacophore. J. Am. Chem. Soc. 2014, 136, 5257–5260.
- Vázquez, K.; Espinosa-Bustos, C.; Soto-Delgado, J.; Tapia, R.A.; Varela, J.; Birriel, E.; Segura, R.; Pizarro, J.; Cerecetto, H.; González, M. New aryloxy-quinone derivatives as potential anti-Chagasic agents: Synthesis, trypanosomicidal activity, electrochemical properties, pharmacophore elucidation and 3D-QSAR analysis. RSC Adv. 2015, 5, 65153–65166.
- Bennett, B.C.; Wan, Q.; Ahmad, M.F.; Langan, P.; Dealwis, C.G. X-ray structure of the ternary MTX NADPH complex of the anthrax dihydrofolate reductase: A pharmacophore for dual-site inhibitor design. J. Struct. Biol. 2009, 166, 162–171.
- François, P.; Hakim, V. Design of genetic networks with specified functions by evolution in silico. Proc. Natl. Acad. Sci. USA 2004, 101, 580–585.
- Fischer, B.; Fukuzawa, K.; Wenzel, W. Receptor-specific scoring functions derived from quantum chemical models improve affinity estimates for in-silico drug discovery. Proteins Struct. Funct. Bioinform. 2008, 70, 1264–1273.
- Khatami, S.G.; Mubeen, S.; Hofmann-Apitius, M. Data science in neurodegenerative disease: Its capabilities, limitations, and perspectives. Curr. Opin. Neurol. 2020, 33, 249.
- Ferro, M.P.; Heilshorn, S.C.; Owens, R.M. Materials for blood brain barrier modeling in vitro. Mater. Sci. Eng. R Rep. 2020, 140, 100522.
- May, J.-N.; Golombek, S.K.; Baues, M.; Dasgupta, A.; Drude, N.; Rix, A.; Rommel, D.; von Stillfried, S.; Appold, L.; Pola, R. Multimodal and multiscale optical imaging of nanomedicine delivery across the blood-brain barrier upon sonopermeation. Theranostics 2020, 10, 1948.
- Juthani, R.; Madajewski, B.; Yoo, B.; Zhang, L.; Chen, P.-M.; Chen, F.; Turker, M.Z.; Ma, K.; Overholtzer, M.; Longo, V.A. Ultrasmall Core-Shell Silica Nanoparticles for Precision Drug Delivery in a High-Grade Malignant Brain Tumor Model. Clin. Cancer Res. 2020, 26, 147–158.
- Blauwendraat, C.; Heilbron, K.; Vallerga, C.L.; Bandres-Ciga, S.; von Coelln, R.; Pihlstrøm, L.; Simón-Sánchez, J.; Schulte, C.; Sharma, M.; Krohn, L. Parkinson’s disease age at onset genome-wide association study: Defining heritability, genetic loci, and α-synuclein mechanisms. Mov. Disord. 2019, 34, 866–875.
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102.
- Ciani, M.; Benussi, L.; Bonvicini, C.; Ghidoni, R. Genome Wide Association Study and Next Generation Sequencing: A glimmer of light towards new possible horizons in Frontotemporal Dementia research. Front. Neurosci. 2019, 13, 506.
- Ibanez, L.; Farias, F.H.; Dube, U.; Mihindukulasuriya, K.A.; Harari, O. Polygenic risk scores in neurodegenerative diseases: A review. Curr. Genet. Med. Rep. 2019, 7, 22–29.
- Adams, H.H.; Evans, T.E.; Terzikhan, N. The Uncovering Neurodegenerative Insights Through Ethnic Diversity consortium. Lancet Neurol. 2019, 18, 915.
- Mancuso, R.; Fryatt, G.; Cleal, M.E.; Obst, J.; Pipi, E.; Monzon-Sandoval, J.; Winchester, L.; Ribe, E.; Webber, C.; Nevado, A. CSF1R inhibition by JNJ-40346527 alters microglial proliferation and phenotype and results in attenuation of neurodegeneration in P301S mice. Brain 2019, 142, 3243–3264.
- Rahman, S.; Datta, M.; Kim, J.; Jan, A.T. CRISPR/Cas: An intriguing genomic editing tool with prospects in treating neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 96, 22–31.
- Offen, D.; Perets, N.; Betzer, O.; Popovtzer, R.; Shapira, R.; Ashery, U. Targeting damages in the brain: Exosomes derived from MSC present migration and homing abilities to different neurodegenerative and neuropsychiatric locations. Cytotherapy 2019, 21, e6.
- Raikwar, S.P.; Kikkeri, N.S.; Sakuru, R.; Saeed, D.; Zahoor, H.; Premkumar, K.; Mentor, S.; Thangavel, R.; Dubova, I.; Ahmed, M.E. Next generation precision medicine: CRISPR-mediated genome editing for the treatment of neurodegenerative disorders. J. Neuroimmune Pharmacol. 2019, 14, 608–641.
- Aldewachi, H.; Al-Zidan, R.N.; Conner, M.T.; Salman, M.M. High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases. Bioengineering 2021, 8, 30.
- Mittal, R.; Woo, F.W.; Castro, C.S.; Cohen, M.A.; Karanxha, J.; Mittal, J.; Chhibber, T.; Jhaveri, V.M. Organ-on-chip models: Implications in drug discovery and clinical applications. J. Cell. Physiol. 2019, 234, 8352–8380.
- Salman, M.M.; Marsh, G.; Kusters, I.; Delincé, M.; Di Caprio, G.; Upadhyayula, S.; de Nola, G.; Hunt, R.; Ohashi, K.G.; Gray, T.; et al. Design and Validation of a Human Brain Endothelial Microvessel-on-a-Chip Open Microfluidic Model Enabling Advanced Optical Imaging. Front. Bioeng. Biotechnol. 2020, 8.
- Chen, J.J. Functional MRI of brain physiology in aging and neurodegenerative diseases. Neuroimage 2019, 187, 209–225.
- Muir, E.R.; Biju, K.; Cong, L.; Rogers, W.E.; Hernandez, E.T.; Duong, T.Q.; Clark, R.A. Functional MRI of the mouse olfactory system. Neurosci. Lett. 2019, 704, 57–61.
- Yan, S.; Zheng, C.; Cui, B.; Qi, Z.; Zhao, Z.; An, Y.; Qiao, L.; Han, Y.; Zhou, Y.; Lu, J. Multiparametric imaging hippocampal neurodegeneration and functional connectivity with simultaneous PET/MRI in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 2440–2452.