Extracellular vesicles (EVs) are cell-derived vesicles important in intercellular communication that play an essential role in host-pathogen interactions, spreading pathogen-derived, as well as host-derived molecules during infection. Pathogens can induce changes in the composition of EVs derived from the infected cells and use them to manipulate their microenvironment and, for instance, modulate innate and adaptive inflammatory immune responses, both in a stimulatory or suppressive manner. Gastric cancer is one of the leading causes of cancer-related deaths worldwide and infection with Helicobacter pylori (H. pylori) is considered the main risk factor for developing this disease, which is characterized by a strong inflammatory component. EVs released by host cells infected with H. pylori contribute significantly to inflammation, and in doing so promote the development of disease.
- Helicobacter pylori
- extracellular vesicles
- outer membrane vesicles
- virulence factors
- inflammation
- gastric cancer
1. Helicobacter pylori, Inflammation and Gastric Cancer
Helicobacter pylori (H. pylori) is a Gram-negative, microaerophilic, spiral bacterium, which colonizes the epithelium of the human stomach [1]. H. pylori infection is one of the most common infectious diseases, affecting approximately 50% of the world’s population [2]. Infection rates differ according to geographic region, remaining higher in developing countries (>85%) [3]. The prevalence of H. pylori infection is associated with low socioeconomic status and poor sanitary conditions, which are considered risk factors [4]. The most likely route of transmission is oral-fecal, especially through contaminated food [5]. In the last three decades, the incidence of H. pylori infection has remained constant or even increased due to population growth, re-infection and increasing antibiotic resistance [6]. H. pylori was described for the first time by Warren and Marshall in biopsies from patients with gastritis and peptic ulceration [7]. Numerous studies have identified H. pylori as the principal etiologic agent associated with the development of chronic gastritis, peptic ulcer disease, mucosa-associated lymphoid tissue lymphoma and gastric cancer [8]. Recently, H. pylori has also been associated with diseases outside the gastrointestinal tract, such as, immune thrombocytopenic-purpura, iron-deficiency anemia, vitamin B12 deficiency, neurodegenerative disorders and metabolic syndrome [9]. H. pylori colonization of the human stomach and its role as a bacterial carcinogen is a complex process involving communication between host cells, the microorganism and the host environment [10]. In the initial phase of infection, H. pylori neutralizes the acidic environment of the stomach by liberating urease, which hydrolyzes urea to generate carbon dioxide and ammonia [11] that neutralize the acidic microenvironment surrounding the bacteria [12]. In addition, ammonia produces various alterations in host cells by affecting vesicular membrane transport, protein synthesis and ATP production, among others [11]. Moreover, urease activity regulates H. pylori-macrophage interactions, by modulating the phagosome pH and megasome formation, which are necessary for H. pylori survival [12]. H pylori has 4–8 flagella and chemotaxis receptors which it uses to colonize the stomach lining and reach epithelial cells [13]. Colonization by H. pylori leads to inflammation and neutrophil infiltration [13][14]. The flagellar filament of H. pylori is mainly composed of the structural proteins FlaA, and FlaB [12]. These flagellins are the first targets of antibodies generated by the host adaptive immune response [13]. However, flagellins, particularly FlaA, are not recognized by toll-like receptor 5 (TLR5) [15][16]. This ability of H. pylori to evade the immune system, contributes to the persistence of the bacteria in the gastric epithelium [13]. H. pylori attaches to the gastric epithelium by producing adhesins, which interact with host cell receptors [12]. These specific interactions protect the bacteria from displacement by forces produced during peristalsis, leading to successful and persistent infection [11][12]. After colonization, virulence factors, such as cytotoxin-associated gene A (CagA) and vacuolating cytotoxin A (VacA), cause further damage to the host epithelium and may increase the risk of gastric disease [10]. The cag pathogenicity island (cagPAI) is a region of the bacterial chromosome, coding CagA and the Cag type IV secretion system (T4SS), which are detectable in more virulent H. pylori strains [10]. Accordingly, the presence of cagPAI has been associated with higher levels of inflammation in the stomach [11]. CagA is an oncoprotein produced in the cytoplasm of the bacteria, which is then injected into host cells via the T4SS [12][17]. Once in the cytoplasm, CagA localizes to the inner surface of the plasma membrane where it is phosphorylated on tyrosine residues by c-SRC and c-ABL kinases [10]. CagA then can modify host cell signaling in multiple ways, such as by activating PI3K/AKT [18], Wnt/-catenin [19] and MAPK/ERK signaling [10]. CagA also modulates cell adhesion and migration by binding to the phosphatase SHP-2 [12][20]. Other effects include the alteration of epithelial cell polarity and the dysregulation of apical-junctional complexes [10][21]. In addition, CagA activates NF-κB-dependent inflammatory signaling and interleukin-8 (IL-8) production in infected gastric cells [10][22]. This leads to recruitment of neutrophils and macrophages, which further increases the release of cytokines, as well as reactive oxygen species (ROS) and growth factors[23][22]. This microenvironment results in chronic inflammation, which promotes tumorigenesis [22]. VacA contains p33 and p55 protein subunits, which combine to generate an oligomeric complex [12] that forms selective anion channels in the host cell membrane and can release bicarbonate and organic anions into the host cytoplasm [12][24]. In this way, VacA aids H. pylori survival by inducing a flow of metabolic substrates from the mucosa to the stomach lumen [25]. This produces changes in the permeability of the plasma membrane and disrupts gastric epithelium cell integrity [12]. VacA also forms channels in the membranes of endosomes and mitochondria [24][26]. Moreover, VacA internalizes into cells via endocytosis, affecting endosomal maturation, among others [12][25]. In late endosomal compartments, these anion selective channels cause an increase in osmotic pressure, resulting in the formation of vacuoles [25][26][27]. In mitochondria, VacA induces changes in the mitochondrial membrane potential and release of cytochrome C leading to apoptosis [25][28]. In addition to its cytotoxic properties, VacA acts as an immunosuppressor by inhibiting the development, activation and proliferation of T cells [10][29], as well as preventing antigen presentation by MHC class II molecules in B cells [25]. Moreover, VacA delays phagocytosis, inhibiting phagosome maturation in macrophages [25][30]. All H. pylori strains contain genes coding for VacA; however, there are multiple alleles that confer variations in vacuolation capacity [10][31]. Studies reveal that VacA in allelic variant s1/m1 and s1/m2 strains is associated with more severe chronic inflammation than for other genotypes [25]. H. pylori is an established group 1 carcinogen and presence of the bacteria is therefore linked to carcinogenesis [32]. Stomach or gastric cancer is a multifactorial and heterogeneous disease, whereby gastric adenocarcinoma (glandular origin) represents the most common histological type (~90%) of all neoplasms originating in the stomach [33][34]. Other less common variants of gastric cancer include lympho-proliferative, mesenchymal and neuroendocrine tumors [33]. Gastric adenocarcinoma is preceded by progressive changes in the gastric mucosa, beginning with non-atrophic gastritis, triggered primarily by H. pylori, followed by multifocal atrophic gastritis, which advances to intestinal metaplasia, dysplasia, and finally invasive carcinoma [35][36]. This cascade of precancerous lesions describes a human model of intestinal-type gastric carcinogenesis, which is the major gastric adenocarcinoma subtype and represents 60% of all cases [34][37]. This intestinal-type gastric cancer is characterized by the presence of tumor cells that are arranged in irregular tubular or glandular structures [38]. The oncogenic effects of H. pylori in epithelial cells are attributed either directly to the toxic action of the virulence factors described above, and/or indirectly to inflammatory processes produced by the infection [39]. Gastritis caused by H. pylori infection is characterized by infiltration of the lamina propria with mononuclear leukocytes (chronic inflammation) and polymorphonuclear cells (acute inflammation) [36]. The immune response triggered by infection is both of the innate and adaptive type [36][39]. T helper-1 (Th1) cells and their signature cytokines, IL-1, tumor necrosis factor (TNF) and particularly interferon (IFN), are essential for the development of gastritis [36][40]. Recruited neutrophils and macrophages produce large amounts of ROS and reactive nitrogen species (RNS) [39][41]. This results in an increase in oxidative stress, which causes DNA damage [39]. Loss of normal glandular tissue, or atrophy, is the result of chronic inflammation and tends to be multifocal (multifocal atrophic gastritis) [36]. The increased expression of multiple cytokines by the gastric mucosa accelerates the progression of atrophic changes in cells [39]. In advanced stages of atrophy, intestinal metaplasia occurs, where a phenotypic change in the gastric mucosa is observed [36]. Normal epithelial cells are replaced by cells with an intestinal phenotype [42]. Intestinal metaplasia is considered a condition that predisposes to malignancy [36][42]. Dysplasia is characterized by a neoplastic phenotype, both in cell morphology and architectural organization of the epithelium [36]. The progression from superficial gastritis to an atrophic, metaplastic and finally neoplastic mucosa is associated with the virulence of H. pylori, as well as environmental and host factors [43]. Both gastric mucosal inflammation and H. pylori may cause host cell genomic instability, damage of the DNA mismatch repair system, abnormal DNA methylation, dysregulation of noncoding gene expression, which all contribute to an accumulation of mutations and the loss of normal regulation of cell function [43][44]. Besides participation of the aforementioned H. pylori virulence factors in disease development, the bacteria also liberates outer membrane vesicles (H. pylori-OMVs) [45][46] that participate in such events. Biologically active H. pylori compounds can be introduced by H. pylori-OMVs into host cells where they may alter cell signaling pathways, impair cellular function, promote infection, and regulate immune responses [31][46]. In addition, H. pylori colonizes the epithelium of the stomach and causes pleiotropic changes in the infected cells of the stomach lining. All eukaryotic cells release host-derived extracellular vesicles (EVs) [47], and after infection by a pathogen these EVs may change in number, composition, or both. The details of these changes and, as a consequence, the biological effects such EVs may have in recipient cells are just beginning to be unraveled. As for H. pylori infection, little is known in this respect. However, the available evidence suggests they are important in this context. Thus, in the rest of this review we will elaborate on the contribution of H. pylori infected host-derived EVs and H. pylori-OMVs to the genesis and progression of gastric cancer.
2. What Are Extracellular Vesicles?
Extracellular vesicles (EVs) are membranous vesicles derived from cells that are important in local and distant intercellular communication, although initially such vesicles were thought to represent a mechanism by which cells eliminate waste products. All human cells that have been investigated are able to secrete EVs. Furthermore, this is a highly conserved process throughout evolution, from bacteria, protozoa, fungi, and plants to animals [48][49][50]. EVs were discovered in sheep reticulocytes, during their differentiation process. Johnstone’s group reported that immature sheep reticulocytes released most of their transferrin receptor in association with small membrane vesicles, through fusion of multi-vesicular bodies (MVBs) with the cell membrane [51][52]. Subsequently, it was shown that reticulocytes from other species also release EVs, which, in addition to transferrin receptors, also contain other proteins, such as acetylcholinesterase [52][53]. Although in the context of reticulocyte development it is clear why EVs were considered a means by which cells eliminated unwanted molecules, nowadays their role is viewed as being much more complex.
3. Extracellular Vesicles in Host–Bacteria Interactions
3.1. Host-Derived Extracellular Vesicles
3.2. Outer Membrane Vesicles in Host-Bacteria Interactions
4. Extracellular Vesicles from Helicobacter pylori-Infected Host Cells. Mechanisms of Action Associated Primarily with Gastric Cancer Development
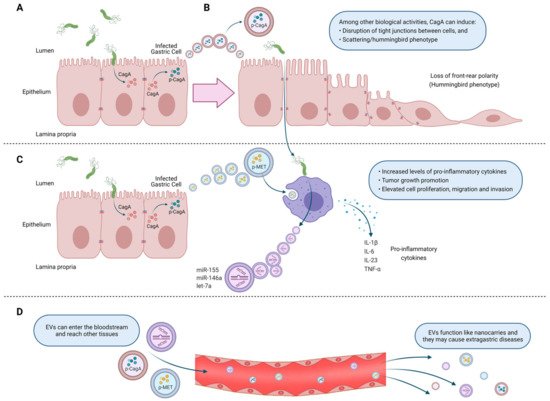
5. Outer Membrane Vesicles from Helicobacter pylori
H. pylori, like many of the Gram-negative bacteria mentioned above, secrete outer membrane vesicles (OMVs) derived from the bacterial outer membrane. These vesicles ranging from 20–450 nm size participate in the communication between bacteria as well as with the environment and, therefore, are important players in bacterial pathogenesis [147][148][149]. It has been reported that H. pylori releases OMVs both in vitro and in vivo [150][151][152][153]. OMVs secreted from H. pylori (H. pylori-OMVs) retain many of the surface molecules of the bacteria, such as LPS, peptidoglycan and outer membrane proteins [147]. Additionally, H. pylori-OMVs contain cytoplasmic molecules, such as proteins associated with translation and virulence factors [154]. Olofsson et al. (2010), determined that H. pylori-OMVs phospholipid composition is similar to that of the bacteria outer membrane and includes phosphatidylglycerol, cardiolipin, lyso-phophatidylethanolamine, phosphatidylethanolamine and cholesterol. However, some components are enriched in the OMVs compared to the bacterial outer membrane, as is the case for the protease HtrA [155]. Mass spectrometry analysis revealed that 77% of the bacterial outer membrane proteins could also be detected in H. pylori-OMVs. Moreover, several proteins associated with H. pylori virulence, such as the urease subunits, VacA, CagA, BabA and SabA adhesins were identified (Figure 2). In addition, glutamyl transpeptidase, the protease HtrA, as well as the cytoplasmic proteins GroEL, catalase, metabolic and ribosomal proteins were also identified in H. pylori-OMVs [155]. Proteomics analysis of OMVs from H. pylori identified a subset of proteins that are not detected in their parent bacterium, suggesting that there is a mechanism for cargo selection [149]. In addition, the size, protein composition and cargo selection for OMVs depended on the stage of H. pylori growth [149]. In this study, H. pylori-OMVs were isolated and purified from bacterial cultures in early, late and stationary growth phase. The results show that H. pylori-OMVs isolated from bacteria in the late and stationary phases of growth are smaller but more abundant than H. pylori-OMVs from an early phase bacteria. Furthermore, OMVs isolated from H. pylori in early growth phase are enriched in metabolic proteins and virulence factors, such as VacA, urease and CagA compared to OMVs from later stage bacteria [149]. On the other hand, the environmental conditions and nutrient availability can also affect the composition of H. pylori-OMVs. When H. pylori was cultured on Brucella broth or blood agar, the derived OMVs showed a different protein pattern [155]. In addition, proteomic analysis revealed that the number of OMV associated proteins may vary between H. pylori strains J99 and NCTC 11637 [154]. However, the mechanisms that produce these differences are unknown. According to epidemiological studies, there is an association between H. pylori infection and iron metabolism. Therefore, an unbalanced iron metabolism could affect the outcome of H. pylori infection [156][157][158]. Interestingly, iron availability affects the composition of H. pylori-OMVs. In particular, Keenan et al. (2008), determined that H. pylori-OMVs obtained from iron-containing media are LPS enriched, whereas H. pylori-OMVs from bacteria grown under iron-limiting conditions have less and shorter LPS [159]. Furthermore, iron deficiency causes a decrease in bacterial growth, but does not reduce the release of OMVs. However, OMVs released under iron deficient conditions contain less VacA, which is consistent with the absence of vacuolation observed in HEp-2 cells after incubation with these OMVs [160].
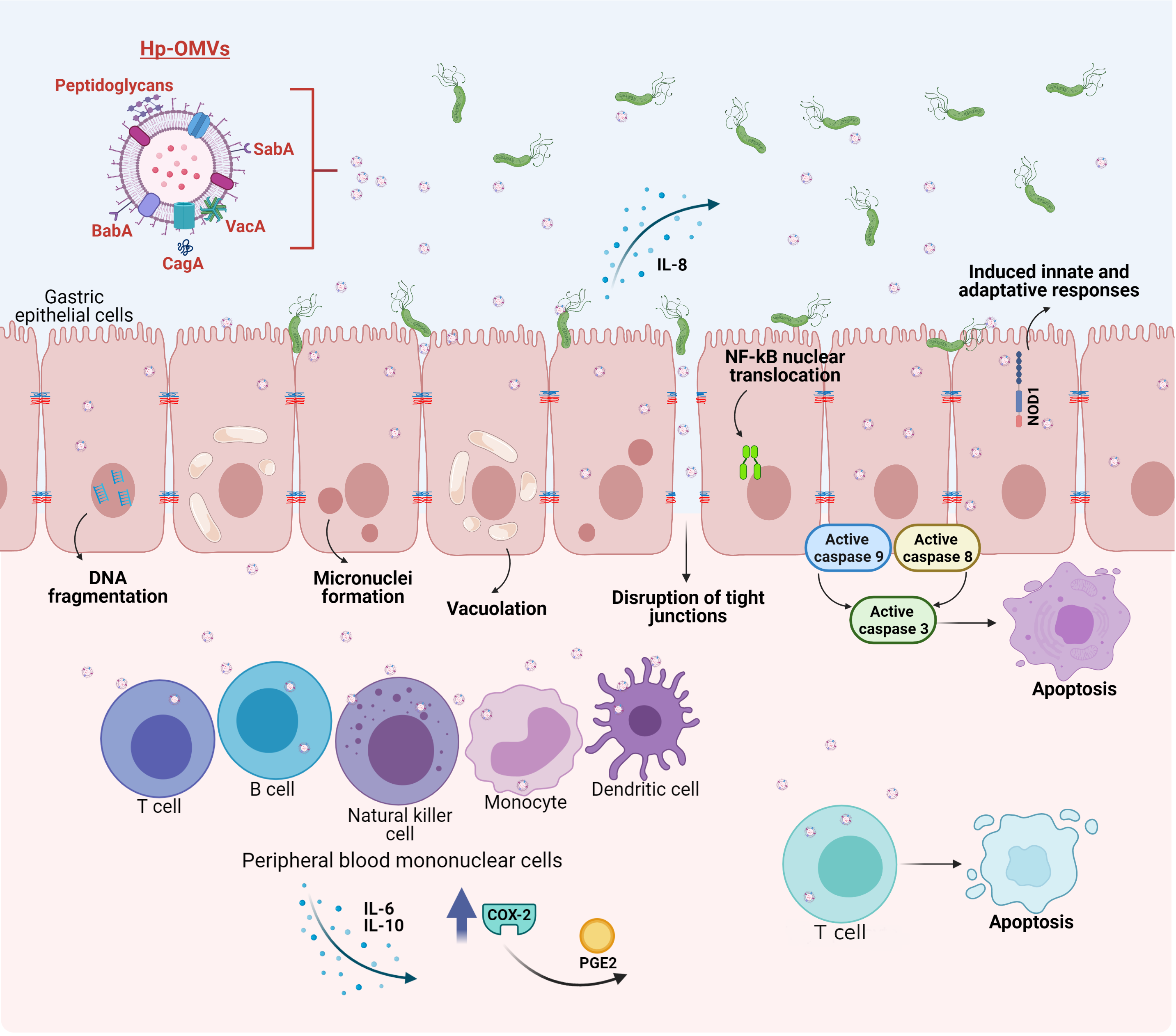
Figure 2. Biological effects of outer membrane vesicles released by H. pylori (H. pylori-OMVs) on host cells. SabA and BabA present in H. pylori-OMVs promote adherence to the gastric mucosa. Internalization of H. pylori-OMVs by epithelial cells leads to DNA fragmentation and activation of caspases-8, 9 and 3. H. pylori-OMVs induce micronuclei formation and vacuoles mediated by VacA. CagA promotes the disruption of tight junctions between cells. Moreover, H. pylori-OMVs induce NF-B nuclear translocation and increase IL-8 production. Peptidoglycans from H. pylori-OMVs, stimulate the cytosolic nucleotide binding oligomerization domain 1 (NOD1) response. In peripheral blood mononuclear cells, H. pylori-OMVs stimulate proliferation and the release of IL-6 and IL-10. H. pylori-OMVs also induce the expression of cyclo-oxygenase-2 (COX-2) and the production of prostaglandin E2 (PGE2). Alternatively, the presence of VacA induces apoptosis in T cells.
OMV formation initiates when protrusions are generated from the outer membrane of Gram-negative bacteria that then pinch off, capturing some of the periplasm content [155]. The ability of H. pylori-OMVs to elicit a response in the host depends on uptake and the entry into epithelial cells [148]. The internalization of OMVs secreted by H. pylori occurs primarily through clathrin-dependent endocytosis and, to a lesser extent, through clathriin-independent mechanisms [161][162]. Concerning cholesterol-dependent mechanisms for internalization, contradictory evidence has been obtained [161][162]. On the other hand, Parker et al. (2010) showed that within 20 min of stimulation of AGS cells with H. pylori-OMVs, all associated OMVs localized within the cells, and that uptake occurred at a higher rate in the presence of VacA. Additionally, the authors observed that H. pylori-OMV uptake is inhibited by addition of LPS [163]. More recently, Turner et al. (2018), demonstrated that the size of OMVs determines the endocytic entry mechanism. A heterogenous sized H. pylori-OMV population results in uptake by micropinocytosis, clathrin- and caveolin-mediated endocytosis. Generally, smaller OMVs (20–100 nm) prefer caveolin-mediated internalization, whereas larger OMVs (90–450 nm) seem to prefer clathrin- and dynamin-mediated endocytosis [148]. Interestingly, larger OMVs contain more and a wider range of proteins than smaller OMVs. For example, large OMVs, but not small OMVs contain BabA and SabA adhesins [148]. However, both populations have virulence factors, such as urease and VacA. Thus, OMV size determines the role these vesicles play in pathogenesis [148].
Accordingly, H. pylori-OMVs transport on their surface or inside, several virulence factors, such as VacA and CagA, and are rapidly internalized by gastric epithelial cells [93][156][163]. Additionally, both the adhesins BabA and SabA were also found on the OMV surface and shown to be biologically active as mediators of adhesion to the human gastric mucosa. There is a high variability between strains in terms of OMV protein profiles. Such variations also affect VacA presence [93][164]. VacA was identified using specific antibodies in H. pylori-OMVs present in gastric biopsies and H. pylori cultures. In addition, VacA was shown to be biologically active in OMVs, since vacuolation was observed after incubation of HEp-2 cells with OMVs from H. pylori 60190 (Figure 2) [151]. Additionally, VacA is internalized and can be observed intracellularly in epithelial cells when these are incubated with either the free form or bound to OMVs. Ricci et al. (2005), showed that approximately 75% of total VacA is released in the free form and the rest is in OMVs. Although both soluble VacA and OMV VacA can induce vacuolization, the OMV VacA is less effective in this respect and may perhaps play a different role [165].
Another important virulence factor identified in OMVs is urease. Proteomic analysis revealed that UreA, a urease catalytic subunit, was present in H. pylori-OMVs isolated from H. pylori strain 26695 and CCUG 17875. Moreover, in AGS cells treated with H. pylori-OMVs from the strain 26695, UreA localized to the cytoplasm and nucleus. Therefore, OMVs can transfer UreA to gastric cells. In addition, the nuclear targeting of UreA lead to morphological changes, suggesting that UreA has functions that are independent of its role as an enzyme [166]. Consistent with this notion, recent studies showed that urease also functions as a ligand for TLR2 to induce stabilization of hypoxia-inducible factor-1 [167].
Catalase (KatA) is a virulence factor that promotes bacterial survival by eliminating hydrogen peroxide and hypochlorite generated by immune cells [168]. Notably, catalase was identified in H. pylori-OMVs at a 7-fold higher concentration than in H. pylori itself. Thus, H. pylori-OMVs, are endowed with a strong anti-oxidant capacity that surrounds the bacteria and protects against oxidative damage produced by the immune system [168].
H. pylori like many bacteria, forms biofilms, which contain extracellular matrix components, including exo-polysaccharides, proteins, lipids and DNA. Moreover, biofilm formation is important for survival and successful infection by many pathogenic bacteria. Interestingly, OMVs are detected in the matrix of H. pylori biofilms and the production of H. pylori-OMVs was found to strongly correlate with biofilm formation [169]. Therefore, another role of OMVs may be to promote H. pylori biofilm formation and thereby enhance bacterial survival as well as favor infection of the stomach epithelium.
6. Biological Effects on Host Cells of Outer Membrane Vesicles Released by Helicobacter pylori. Mechanisms of Action Associated with Gastric Cancer Development
OMVs from H. pylori have many effects, both in vitro and in vivo [170]. In particular, H. pylori-OMVs have been shown to induce micronuclei formation in gastric AGS cells, indicating genomic damage, apparently mediated by VacA. In the same gastric epithelial cell line, internalization of H. pylori-OMVs lead to the formation of large vacuoles, as early as 4 h after incubation (Figure 2). Additionally, disruption of vacuole integrity and reduction in cellular glutathione were observed and restoring glutathione levels by the addition of glutathione ester in vitro to AGS cells prevented micronuclei formation caused by H. pylori-OMV stimulation, suggesting a role for oxidative stress in the infection process [153]. As mentioned previously, H. pylori-OMVs also contain active CagA, which appears to promote the disruption of tight junctions between epithelial cells (Figure 2), given that CagA from H. pylori-OMVs localizes in close proximity of zona occludens-1 protein junctions, which correlates with a disorganized and diffuse zona occludens-1 pattern after stimulation by H. pylori-OMVs. Moreover, CagA in H. pylori-OMVs enhances histone H1 affinity for ATP, which may modulate gene transcription, although this point was not addressed in the study [171].
Adhesins important for H. pylori adherence to the human gastric epithelium, such as SabA and BabA, are present in H. pylori-OMVs, and are biologically active, as they promote adherence between OMVs isolated in vitro, as well as to the human gastric mucosa in biopsies. Moreover, pre-incubation of H. pylori-OMVs with Leb-receptor or sLex receptor conjugates before adding OMVs to the human gastric mucosa from biopsies, prevented H. pylori-OMV binding, suggesting that SabA and BabA are important for adherence to the gastric mucosa [155]. The effect of H. pylori-OMVs on epithelial cell proliferation appears to be dose dependent. After 24 h of stimulation, low OMV doses increased AGS cell proliferation up to 30%; alternatively, at high doses, approximately 50% growth arrest and a decrease in proliferation is observed. In addition, high doses of H. pylori-OMVs increase toxicity and IL-8 production [163][172]. The decrease in cell viability of gastric epithelial cells after stimuli with H. pylori-OMVs was also corroborated by others [173]. Other effects in AGS cells observed in this study included vacuolation and apoptosis. Specifically, DNA fragmentation and activation of the caspases-8, 9 and 3 were observed but these events occurred in the absence of cytochrome C release (Figure 2) [173].
Less is known about the effects of H. pylori-OMVs on cells of the host immune system; however recent evidence shows that they have opposite effects. For instance, treatment with H. pylori-OMVs stimulate proliferation and the release of IL-6 and 10 from peripheral blood mononuclear cells [93]. Alternatively, H. pylori-OMVs induce apoptosis in Jurkat T cells (immortalized human T cells) and in naïve CD4+ cells [93]. H. pylori-OMVs also induce the expression of cyclo-oxygenase-2 in peripheral blood mononuclear cells, and as a consequence, H. pylori-OMVs can inhibit human T cell proliferation, via prostaglandin-E2. Induction of IL-10 was also observed in peripheral blood mononuclear cells (Figure 2) [174].
H. pylori-OMVs induced, in a dose-dependent manner, IL-8 in gastric AGS cells, and IL-6 as well as TNF in mouse macrophages. In vivo, oral and intraperitoneal administration of H. pylori-OMVs in mice induced IFN-α, IL-17 and IL-4 expression, as well as increased immunoglobulin G generation. In vivo, H. pylori-OMVs targeted stomach epithelial cells in mice. Thus, H. pylori-OMVs induce immune responses both in vitro and in vivo. Since, as mentioned before, H. pylori-OMVs are abundant in the stomach of gastric cancer patients, these can infiltrate the gastric mucosa to induce inflammation, and potentially promote gastric cancer [142].
H. pylori-OMVs also contain peptidoglycans, which stimulate the cytosolic nucleotide binding oligomerization domain 1 (NOD1) response in epithelial cells (Figure 2). In addition, H. pylori-OMVs administered intra-gastrically induced innate and adaptive immune responses in mice, via NOD1. Specifically, H. pylori-OMVs induce NF-κB reporter activity and nuclear translocation of the NF-κB p65 subunit, in addition to IL-8 production (Figure 2) [147]. IL-8 and IL-6 are pro-inflammatory cytokines that prolong inflammation, while IL-10 is an anti-inflammatory cytokine, which has a suppressive function on cytokine secretion. Thus, H. pylori-OMVs can elicit opposing effects in the immune system, by increasing or decreasing the proliferation of immune cells, as well as by elevating both, pro- and anti-inflammatory cytokines. Further research is required to determine how H. pylori-OMVs contribute to the development of gastric cancer associated with H. pylori infection.
7. Helicobacter pylori-Outer Membrane Vesicles and Extracellular Vesicles from Helicobacter pylori Infected-Host Cells in Vaccine Development and Biomarker Research
Recent applications of OMV and EV research include vaccine development and biomarker identification [175][176]. Currently, the first line of treatment against H. pylori is antibiotic combination therapy. However, in the last decades resistance to antibiotics has increased [177]. Therefore, new alternative forms of therapy are being developed, and vaccines have emerged as a potential candidate [178].
Vaccines developed against the bacterial outer membrane have been used to induce protection against various pathogens. Hence, the use of OMVs for the same purpose has recently been explored. The first challenge in developing a vaccine targeting H. pylori-OMVs, is the method used for isolating large quantities of OMVs. In this respect, Turner et al. (2015), described that the TolB protein is key to H. pylori membrane integrity and H. pylori-OMVs from a DtolB mutant strain increased the production of H. pylori-OMVs that were more immunogenic [179]. Keenan et al. (2000) evaluated immunogenicity of H. pylori-OMVs in a murine model of immune protection and such effects were attributed to increased serum immunoglobulin G antibody against lipoprotein 20 [175]. Liu et al. (2019), tested the immune protective response of H. pylori-OMVs obtained from the gerbil-adapted H. pylori strain 7.13 using a mouse model. The results showed that H. pylori-OMVs generate a strong humoral and mucosal immune response without inflammation that reduces H. pylori load, suggesting a potential use as a treatment [178]. Subsequently, the combination of H. pylori-OMVs with outer membrane proteins or whole cell vaccines, two commonly studied approaches against H. pylori, confirmed that H. pylori-OMVs from H. pylori strain 7.13 were effective as an adjuvant. The analysis demonstrated that vaccines with H. pylori-OMVs induced a Th2 and Th17 biased immunity, increasing cell mediated and humoral immunity, which reduces H. pylori colonization in a mouse model. Consequently, H. pylori-OMVs are effective adjuvants for these types of vaccines [180].
The high death rate from gastric cancer is due to the fact that most patients with the disease are not diagnosed until they exhibit symptoms. This makes it difficult to detect gastric cancer at an early stage [181]. Since the infection with H. pylori is associated with the development of gastric precancerous lesions and gastric cancer, its detection is essential for early diagnosis. Current H. pylori diagnosis involves both invasive and non-invasive techniques. The invasive approach requires obtaining an endoscopic biopsy, which can be analyzed enzymatically using the rapid urease test or histologically with specific antibodies or by PCR. Among the techniques for processing gastric biopsies, PCR appears to be a more sensitive method compared to the others [182]. Progress has been made in obtaining endoscopic images through the development of linked color imaging (LCI) and blue laser imaging (BLI), whereby sensitivity and specificity are on the order of 83.8–85.4% and 79.5–99.5%, respectively [183]. In addition, artificial intelligence has been used to analyze the presence of H. pylori in images obtained by LCI [184]. On the other hand, non-invasive tests include the urea breathing test (UBT), serology and the H. pylori stool antigen test. For the UBT, patients ingest labeled 13C-urea as a substrate for the bacterial urease [185][186]. The 13C-labeled carbon dioxide produced by the reaction is detected in the patient’s breath by mass spectrometry or using an isotope-selective infrared spectroscope [185]. In recent years, the development of portable mass spectrometers has reduced equipment size and the cost of the analysis [187]. Serology is useful mostly for screening and in epidemiological studies [188]. To date, none of the current H. pylori diagnostic options can be considered a gold standard individually. For instance, besides its invasiveness, endoscopic imaging and biopsies require specialized equipment and medical staff. Regarding the non-invasive methods, the sensitivity and specificity still needs to be improved, although values lie between 93–96% and 94–97% for the UBT and the fecal antigen test, respectively [188]. Of note, efforts have been made in the area of next generation sequencing to permit the detection of H. pylori mutations that confer resistance to the most commonly used antibiotics and coincide phenotypically with antibiotic susceptibility [188]. On the other hand, although it is well known that the serological test does not necessarily detect a current infection, an attempt has been made to diagnose an active infection by detecting seropositivity for 13 H. pylori proteins by multiplex. This study concludes that seropositivity to 2 of the proteins VacA, GroEl, HcpC and HP1564 is sufficient to identify an ongoing H. pylori infection [189]. The sensitivity limitations and the invasive nature of most diagnostic methods, together with the lack of a gold standard method, make it necessary to develop more sensitive and specific non-invasive tests for the detection of H. pylori infection.
EVs can potentially be used as biomarker carriers, since their content is protected from degradation in the bloodstream by their lipid membrane. These EVs can be isolated from blood or other biological fluids, such as urine, gastric juice or saliva, among others, to identify molecules associated with different diseases. EVs exhibit variations in the molecular profile in healthy subjects compared to patients with diverse diseases, and such information can be exploited for diagnosis [190]. Indeed, as mentioned before, during H. pylori infection, CagA is detected in EVs from patient sera [135]. However, there are not many studies that identify H. pylori-OMVs in circulation, mainly because it is still very difficult to differentiate pathogen-derived OMVs from EVs produced by host cells. However, some reports have revealed the presence of OMVs in circulation [130][191]. Current improvements in methods for the isolation of EVs and pathogen-derived OMVs, along with the identification of molecules that serve as biomarkers, suggest that EVs could be used as a diagnostic tool for the detection of H. pylori infection and preneoplastic gastric lesions in the near future.
8. Conclusions
During H. pylori infection, host cells release EVs carrying an altered profile of host molecules, and even pathogen-derived proteins. These H. pylori-infected host cell EVs can have a variety of effects on gastric epithelial cells, gastric cancer cells and macrophages, including changes in epithelial cell morphology and increased release of cytokines by immune cells. In addition, during H. pylori infection, the bacteria releases OMVs, bearing pathogen-derived molecules, including the virulence factors CagA, VacA, Urease, LPS, SabA, BabA, among many others, that induce changes in epithelial cells, including vacuolation, micronuclei formation, disruption of tight junctions between epithelial cells, and either cell proliferation or growth arrest, depending on the OMV dose. The effects of H. pylori-OMVs on the immune system have been found to be contradictory, as they promote or inhibit proliferation of certain immune cells and increase pro-inflammatory and anti-inflammatory cytokine release. Overall, H. pylori-infected host cell EVs and H. pylori-OMVs can promote cellular changes that tend to favor the development and/or progression of gastric cancer. In addition, H. pylori-OMVs are being evaluated for use in the development of vaccines against H. pylori development and so far, show promising results. Finally, EVs have proven to be useful for diagnosis, as they can be isolated from various biological fluids, such as blood, urine, saliva and gastric juice, among others. Improvements in the purification of EVs and in the identification of biomarkers for detection of H. pylori infection, as well as preneoplastic gastric lesions, hold considerable promise for use in the diagnosis and prevention of gastric cancer. Future studies on EVs/OMVs in H. pylori infection should focus on the development of vaccines against H. pylori and early diagnosis of H. pylori infection along with preneoplastic gastric lesions, in order to improve the prevention of gastric cancer and the survival of gastric cancer patients.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22094823
References
- Milad Azami; Marzieh Parizad Nasirkandy; Akram Mansouri; Zahra Darvishi; Shoboo Rahmati; Ghobad Abangah; Hamid Reza Dehghan; Milad Borji; Shamsi Abbasalizadeh; Global Prevalence of Helicobacter pylori Infection in Pregnant Women: A Systematic Review and Meta-analysis Study. International Journal of Women's Health and Reproduction Sciences 2016, 5, 30-36, 10.15296/ijwhr.2017.06.
- Sheila E. Crowe; Helicobacter pylori Infection. New England Journal of Medicine 2019, 380, 1158-1165, 10.1056/nejmcp1710945.
- Y. Hu; J.-H. Wan; X.-Y. Li; D. Y. Graham; N.-H. Lu; Systematic review with meta-analysis: the global recurrence rate of Helicobacter pylori. Alimentary Pharmacology & Therapeutics 2017, 46, 773-779, 10.1111/apt.14319.
- Mārcis Leja; Anthony Axon; Hermann Brenner; Epidemiology of Helicobacter pylori infection. Helicobacter 2016, 21, 3-7, 10.1111/hel.12332.
- Mohammad Zamani; Amin Vahedi; Zahra Maghdouri; Javad Shokri-Shirvani; Role of food in environmental transmission of Helicobacter pylori. Caspian journal of internal medicine 1969, 8, 146-152, 10.22088/cjim.8.3.146.
- I. Thung; H. Aramin; V. Vavinskaya; S. Gupta; J. Y. Park; S. E. Crowe; M. A. Valasek; Review article: the global emergence ofHelicobacter pyloriantibiotic resistance. Alimentary Pharmacology & Therapeutics 2015, 43, 514-533, 10.1111/apt.13497.
- BarryJ Marshall; J.Robin Warren; UNIDENTIFIED CURVED BACILLI IN THE STOMACH OF PATIENTS WITH GASTRITIS AND PEPTIC ULCERATION. The Lancet 1984, 323, 1311-1315, 10.1016/s0140-6736(84)91816-6.
- Peter Nagy; Saga Johansson; Michael Molloy-Bland; Systematic review of time trends in the prevalence of Helicobacter pylori infection in China and the USA. Gut Pathogens 2016, 8, 1-14, .
- Antonietta Gerarda Gravina; Rocco Maurizio Zagari; Cristiana De Musis; Lorenzo Romano; Carmelina Loguercio; Marco Romano; Helicobacter pyloriand extragastric diseases: A review. World Journal of Gastroenterology 2018, 24, 3204-3221, 10.3748/wjg.v24.i29.3204.
- Manuel Amieva; Richard M. Peek; Pathobiology of Helicobacter pylori–Induced Gastric Cancer. Gastroenterology 2015, 150, 64-78, 10.1053/j.gastro.2015.09.004.
- Bruna M. Roesler; Elizabeth M.A. Rabelo-Gonçalves; José M.R. Zeitune; Virulence Factors of Helicobacter pylori: A Review. Clinical Medicine Insights: Gastroenterology 2013, 7, 9-17, 10.4137/cgast.s13760.
- Cheng-Yen Kao; Bor-Shyang Sheu; Jiunn-Jong Wu; Helicobacter pylori infection: An overview of bacterial virulence factors and pathogenesis. Biomedical Journal 2016, 39, 14-23, 10.1016/j.bj.2015.06.002.
- Haiying Gu; Role of Flagella in the Pathogenesis of Helicobacter pylori. Current Microbiology 2017, 74, 863-869, 10.1007/s00284-017-1256-4.
- Eitaro Aihara; Chet Closson; Andrea L. Matthis; Michael A. Schumacher; Amy C. Engevik; Yana Zavros; Karen M. Ottemann; Marshall H. Montrose; Motility and Chemotaxis Mediate the Preferential Colonization of Gastric Injury Sites by Helicobacter pylori. PLOS Pathogens 2014, 10, e1004275, 10.1371/journal.ppat.1004275.
- Jerneja Mori; Tanja Vranac; Boštjan Smrekar; Maja Černilec; Vladka Čurin Šerbec; Simon Horvat; Alojz Ihan; Mojca Benčina; Roman Jerala; Chimeric flagellin as the self-adjuvanting antigen for the activation of immune response against Helicobacter pylori. Vaccine 2012, 30, 5856-5863, 10.1016/j.vaccine.2012.07.011.
- Jingyi Yang; Huimin Yan; TLR5: beyond the recognition of flagellin. Cellular & Molecular Immunology 2017, 14, 1017-1019, 10.1038/cmi.2017.122.
- Nicole Tegtmeyer; Judith Lind; Benedikt Schmid; Steffen Backert; Helicobacter pylori CagL Y58/E59 Mutation Turns-Off Type IV Secretion-Dependent Delivery of CagA into Host Cells. PLOS ONE 2014, 9, e97782, 10.1371/journal.pone.0097782.
- Na Li; Bin Tang; Yin-Ping Jia; Pan Zhu; Yuan Zhuang; Yao Fang; Qian Li; Kun Wang; Wei-Jun Zhang; Gang Guo; et al. Helicobacter pylori CagA Protein Negatively Regulates Autophagy and Promotes Inflammatory Response via c-Met-PI3K/Akt-mTOR Signaling Pathway. Frontiers in Cellular and Infection Microbiology 2017, 7, 417, 10.3389/fcimb.2017.00417.
- Xin Yong; Bo Tang; Yu-Feng Xiao; Rui Xie; Yong Qin; Gang Luo; Chang-Jiang Hu; Hui Dong; Shi-Ming Yang; Helicobacter pylori upregulates Nanog and Oct4 via Wnt/β-catenin signaling pathway to promote cancer stem cell-like properties in human gastric cancer. Cancer Letters 2016, 374, 292-303, 10.1016/j.canlet.2016.02.032.
- Geneviève Coulombe; Nathalie Rivard; New and Unexpected Biological Functions for the Src-Homology 2 Domain-Containing Phosphatase SHP-2 in the Gastrointestinal Tract. Cellular and Molecular Gastroenterology and Hepatology 2015, 2, 11-21, 10.1016/j.jcmgh.2015.11.001.
- Lydia E Wroblewski; M Blanca Piazuelo; Rupesh Chaturvedi; Michael Schumacher; Eitaro Aihara; Rui Feng; Jennifer M Noto; Alberto Delgado; Dawn A Israel; Yana Zavros; et al. Helicobacter pyloritargets cancer-associated apical-junctional constituents in gastroids and gastric epithelial cells. Gut 2014, 64, 720-730, 10.1136/gutjnl-2014-307650.
- Rebecca J. Gorrell; Jyeswei Guan; Yue Xin; Mona Anoushiravani Tafreshi; Melanie L. Hutton; Michael A. McGuckin; Richard L. Ferrero; Terry Kwok; A novel NOD1- and CagA-independent pathway of interleukin-8 induction mediated by theHelicobacter pyloritype IV secretion system. Cellular Microbiology 2012, 15, 554-570, 10.1111/cmi.12055.
- Langgeng Agung Waskito; Nina R. Salama; Yoshio Yamaoka; Pathogenesis of Helicobacter pylori infection. Helicobacter 2018, 23, e12516, 10.1111/hel.12516.
- Melissa G. Chambers; Tasia M. Pyburn; Christian González-Rivera; Scott E. Collier; İlyas Eli; Calvin K. Yip; Yoshimasa Takizawa; D. Borden Lacy; Timothy L. Cover; Melanie D. Ohi; et al. Structural Analysis of the Oligomeric States of Helicobacter pylori VacA Toxin. Journal of Molecular Biology 2013, 425, 524-535, 10.1016/j.jmb.2012.11.020.
- Nidhi Chauhan; Alfred Chin Yen Tay; Barry J. Marshall; Utkarsh Jain; Helicobacter pyloriVacA, a distinct toxin exerts diverse functionalities in numerous cells: An overview. Helicobacter 2018, 24, e12544, 10.1111/hel.12544.
- Samuel Leslie Palframan; Terry Kwok; Kipros Gabriel; Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori pathogenesis. Frontiers in Cellular and Infection Microbiology 2011, 2, 92, 10.3389/fcimb.2012.00092.
- M. R. Terebiznik; C. L. Vazquez; K. Torbicki; D. Banks; T. Wang; W. Hong; S. R. Blanke; M. I. Colombo; N. L. Jones; Helicobacter pylori VacA Toxin Promotes Bacterial Intracellular Survival in Gastric Epithelial Cells. Infection and Immunity 2006, 74, 6599-6614, 10.1128/iai.01085-06.
- Joachim Rassow; Helicobacter pylori vacuolating toxin A and apoptosis. Cell Communication and Signaling 2010, 9, 26-26, 10.1186/1478-811x-9-26.
- Beate Kern; Utkarsh Jain; Ciara Utsch; Andreas Otto; Benjamin Busch; Luisa Jiménez-Soto; Dörte Becher; Rainer Haas; Characterization ofHelicobacter pylori VacA-containing vacuoles (VCVs), VacA intracellular trafficking and interference with calcium signalling in T lymphocytes. Cellular Microbiology 2015, 17, 1811-1832, 10.1111/cmi.12474.
- Aleksandra Djekic; Anne Müller; The Immunomodulator VacA Promotes Immune Tolerance and Persistent Helicobacter pylori Infection through Its Activities on T-Cells and Antigen-Presenting Cells. Toxins 2016, 8, 187, 10.3390/toxins8060187.
- Shamshul Ansari; Yoshio Yamaoka; Helicobacter pylori Virulence Factors Exploiting Gastric Colonization and its Pathogenicity. Toxins 2019, 11, 677, 10.3390/toxins11110677.
- IARC; Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7-14 June 1994.. IARC monographs on the evaluation of carcinogenic risks to humans 1993, 61, 1-241, .
- Jaffer A. Ajani; Jeeyun Lee; Takeshi Sano; Yelena Y. Janjigian; Daiming Fan; Shumei Song; Gastric adenocarcinoma. Nature Reviews Disease Primers 2017, 3, 17036, 10.1038/nrdp.2017.36.
- Shamshul Ansari; Boldbaatar Gantuya; Vo Phuoc Tuan; Yoshio Yamaoka; Diffuse Gastric Cancer: A Summary of Analogous Contributing Factors for Its Molecular Pathogenicity. International Journal of Molecular Sciences 2018, 19, 2424, 10.3390/ijms19082424.
- Pelayo Correa; A MODEL FOR GASTRIC CANCER EPIDEMIOLOGY. The Lancet 1975, 306, 58-60, 10.1016/s0140-6736(75)90498-5.
- Pelayo Correa Blanca Piazuelo; The Gastric Precancerous Cascade. Journal of Clinical & Experimental Pathology 2012, 3, 2-9, 10.4172/2161-0681.1000147.
- Pekka Laurén; THE TWO HISTOLOGICAL MAIN TYPES OF GASTRIC CARCINOMA: DIFFUSE AND SO-CALLED INTESTINAL-TYPE CARCINOMA. Acta Pathologica Microbiologica Scandinavica 1965, 64, 31-49, 10.1111/apm.1965.64.1.31.
- Junli Ma; Hong Shen; Linda Kapesa; Shan Zeng; Lauren classification and individualized chemotherapy in gastric cancer. Oncology Letters 2016, 11, 2959-2964, 10.3892/ol.2016.4337.
- Sauid Ishaq; Lois Nunn; Helicobacter pylori and gastric cancer: a state of the art review. Gastroenterology and hepatology from bed to bench 2014, 8, S6-S14, .
- Nader Bagheri; Loghman Salimzadeh; Hedayatollah Shirzad; The role of T helper 1-cell response in Helicobacter pylori-infection. Microbial Pathogenesis 2018, 123, 1-8, 10.1016/j.micpath.2018.06.033.
- Osamu Handa; Yuji Naito; Toshikazu Yoshikawa; Redox biology and gastric carcinogenesis: the role ofHelicobacter pylori. Redox Report 2010, 16, 1-7, 10.1179/174329211x12968219310756.
- Veronique Giroux; Anil K. Rustgi; Metaplasia: tissue injury adaptation and a precursor to the dysplasia–cancer sequence. Nature Cancer 2017, 17, 594-604, 10.1038/nrc.2017.68.
- Muhammad Miftahussurur; Yoshio Yamaoka; David Y. Graham; Helicobacter pylorias an oncogenic pathogen, revisited. Expert Reviews in Molecular Medicine 2017, 19, e4-e4, 10.1017/erm.2017.4.
- Alejandra Sandoval-Bórquez; Kathleen Saavedra; Gonzalo Carrasco-Avino; Benjamin Garcia-Bloj; Jacqueline Fry; Ignacio Wichmann; Alejandro H. Corvalán; Noncoding Genomics in Gastric Cancer and the Gastric Precancerous Cascade: Pathogenesis and Biomarkers. Disease Markers 2015, 2015, 1-14, 10.1155/2015/503762.
- Iva Polakovicova; Sofia Jerez; Ignacio A. Wichmann; Alejandra Sandoval-Bórquez; Nicolás Carrasco-Véliz; Alejandro H. Corvalán; Role of microRNAs and Exosomes in Helicobacter pylori and Epstein-Barr Virus Associated Gastric Cancers. Frontiers in Microbiology 2018, 9, 636, 10.3389/fmicb.2018.00636.
- Magdalena Chmiela; Juozas Kupcinskas; Review: Pathogenesis of Helicobacter pylori infection. Helicobacter 2019, 24, e12638, 10.1111/hel.12638.
- X. Delabranche; A. Berger; J. Boisramé-Helms; F. Meziani; Microparticles and infectious diseases. Médecine et Maladies Infectieuses 2012, 42, 335-343, 10.1016/j.medmal.2012.05.011.
- Charlotte Lawson; Dory Kovacs; Elizabeth Finding; Emily Ulfelder; Virginia Luis-Fuentes; Extracellular Vesicles: Evolutionarily Conserved Mediators of Intercellular Communication. The Yale journal of biology and medicine 2017, 90, 481-491, .
- Lisa Brown; Julie M. Wolf; Rafael Prados-Rosales; Arturo Casadevall; Through the wall: extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nature Reviews Genetics 2015, 13, 620-630, 10.1038/nrmicro3480.
- Jeffrey S Schorey; Yong Cheng; Prachi P Singh; Victoria L Smith; Exosomes and other extracellular vesicles in host–pathogen interactions. EMBO reports 2014, 16, 24-43, 10.15252/embr.201439363.
- B T Pan; K Teng; C Wu; M Adam; R M Johnstone; Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes.. Journal of Cell Biology 1985, 101, 942-948, 10.1083/jcb.101.3.942.
- R M Johnstone; M Adam; J R Hammond; L Orr; C Turbide; Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes).. Journal of Biological Chemistry 1987, 262, 9412-9420, 10.1016/s0021-9258(18)48095-7.
- R M Johnstone; J Ahn; A common mechanism may be involved in the selective loss of plasma membrane functions during reticulocyte maturation.. Biomedica biochimica acta 1989, 49, 70-75, .
- Yuana Yuana; Auguste Sturk; Rienk Nieuwland; Extracellular vesicles in physiological and pathological conditions. Blood Reviews 2012, 27, 31-39, 10.1016/j.blre.2012.12.002.
- Ronit Machtinger; Louise C. Laurent; Andrea A. Baccarelli; Extracellular vesicles: roles in gamete maturation, fertilization and embryo implantation. Human Reproduction Update 2015, 22, 182-93, 10.1093/humupd/dmv055.
- Edit I. Buzas; Bence György; György Nagy; András Falus; Steffen Gay; Emerging role of extracellular vesicles in inflammatory diseases. Nature Reviews Rheumatology 2014, 10, 356-364, 10.1038/nrrheum.2014.19.
- Yunmeng Yan; GuangZhen Fu; Yafei Ye; Liang Ming; Exosomes participate in the carcinogenesis and the malignant behavior of gastric cancer. Scandinavian Journal of Gastroenterology 2017, 52, 499-504, 10.1080/00365521.2016.1278458.
- Guillaume Van Niel; Gisela D'Angelo; Graça Raposo; Shedding light on the cell biology of extracellular vesicles. Nature Reviews Molecular Cell Biology 2018, 19, 213-228, 10.1038/nrm.2017.125.
- Valentina R. Minciacchi; Michael R. Freeman; Dolores Di Vizio; Extracellular Vesicles in Cancer: Exosomes, Microvesicles and the Emerging Role of Large Oncosomes. Seminars in Cell & Developmental Biology 2015, 40, 41-51, 10.1016/j.semcdb.2015.02.010.
- Stefania Tavano; Carl-Philipp Heisenberg; Migrasomes take center stage. Nature 2019, 21, 918-920, 10.1038/s41556-019-0369-3.
- Johnny C. Akers; David Gonda; Ryan Kim; Bob S. Carter; Clark C. Chen; Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. Journal of Neuro-Oncology 2013, 113, 1-11, 10.1007/s11060-013-1084-8.
- Nicholas Syn; Lingzhi Wang; Gautam Sethi; Jean-Paul Thiery; Boon-Cher Goh; Exosome-Mediated Metastasis: From Epithelial–Mesenchymal Transition to Escape from Immunosurveillance. Trends in Pharmacological Sciences 2016, 37, 606-617, 10.1016/j.tips.2016.04.006.
- Killian O’Brien; Koen Breyne; Stefano Ughetto; Louise C. Laurent; Xandra O. Breakefield; RNA delivery by extracellular vesicles in mammalian cells and its applications. Nature Reviews Molecular Cell Biology 2020, 21, 585-606, 10.1038/s41580-020-0251-y.
- Tatyana Vagner; Cristiana Spinelli; Valentina R. Minciacchi; Leonora Balaj; Mandana Zandian; Andrew Conley; Andries Zijlstra; Michael R. Freeman; Francesca Demichelis; Subhajyoti De; et al. Large extracellular vesicles carry most of the tumour DNA circulating in prostate cancer patient plasma. Journal of Extracellular Vesicles 2018, 7, 1505403, 10.1080/20013078.2018.1505403.
- America Campos; Carlos Salomon; Rocio Bustos; Jorge Díaz; Samuel Martínez; Veronica Silva; Constanza Reyes; Natalia Díaz-Valdivia; Manuel Varas-Godoy; Lorena Lobos-González; et al. Caveolin-1-containing extracellular vesicles transport adhesion proteins and promote malignancy in breast cancer cell lines. Nanomedicine 2018, 13, 2597-2609, 10.2217/nnm-2018-0094.
- Rachna Manek; Ahmed Moghieb; Zhihui Yang; Dhwani Kumar; Firas Kobessiy; George Anis Sarkis; Vijaya Raghavan; Kevin K.W. Wang; Protein Biomarkers and Neuroproteomics Characterization of Microvesicles/Exosomes from Human Cerebrospinal Fluid Following Traumatic Brain Injury. Molecular Neurobiology 2017, 55, 6112-6128, 10.1007/s12035-017-0821-y.
- Dong-Sic Choi; Dae-Kyum Kim; Yoon-Keun Kim; Yong Song Gho; Proteomics, transcriptomics and lipidomics of exosomes and ectosomes. PROTEOMICS 2013, 13, 1554-1571, 10.1002/pmic.201200329.
- Erik R. Abels; Xandra O. Breakefield; Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cellular and Molecular Neurobiology 2016, 36, 301-312, 10.1007/s10571-016-0366-z.
- Samir El Andaloussi; Imre Mäger; Xandra O. Breakefield; Matthew J. A. Wood; Extracellular vesicles: biology and emerging therapeutic opportunities. Nature Reviews Drug Discovery 2013, 12, 347-357, 10.1038/nrd3978.
- Zhang, Huang-Ge. Emerging Concepts of Tumor Exosome–Mediated Cell-Cell Communication; Springer: New York, 2013; pp. 107-122.
- Guillaume van Niel; Stéphanie Charrin; Sabrina Simoes; Maryse Romao; Leila Rochin; Paul Saftig; Michael S. Marks; Eric Rubinstein; Graça Raposo; The Tetraspanin CD63 Regulates ESCRT-Independent and -Dependent Endosomal Sorting during Melanogenesis. Developmental Cell 2011, 21, 708-721, 10.1016/j.devcel.2011.08.019.
- Kobina Essandoh; Liwang Yang; Xiaohong Wang; Wei Huang; Dongze Qin; Jiukuan Hao; Yigang Wang; Basilia Zingarelli; Tianqing Peng; Guo-Chang Fan; et al. Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2015, 1852, 2362-2371, 10.1016/j.bbadis.2015.08.010.
- Guillaume Van Niel; Graça Raposo; Céline Candalh; Muriel Boussac; Robert Hershberg; Nadine Cerf–Bensussan; Martine Heyman; Intestinal epithelial cells secrete exosome–like vesicles. Gastroenterology 2001, 121, 337-349, 10.1053/gast.2001.26263.
- Sonja I. Buschow; Esther N. M. Nolte-‘T Hoen; Guillaume Van Niel; Maaike S. Pols; Toine Ten Broeke; Marjolein Lauwen; Ferry Ossendorp; Cornelis J. M. Melief; Graça Raposo; Richard Wubbolts; et al. MHC II in Dendritic Cells is Targeted to Lysosomes or T Cell-Induced Exosomes Via Distinct Multivesicular Body Pathways. Traffic 2009, 10, 1528-1542, 10.1111/j.1600-0854.2009.00963.x.
- Kévin Carayon; Karima Chaoui; Elsa Ronzier; Ikrame Lazar; Justine Bertrand-Michel; Véronique Roques; Stéphanie Balor; François Terce; André Lopez; Laurence Salomé; et al. Proteolipidic Composition of Exosomes Changes during Reticulocyte Maturation*. Journal of Biological Chemistry 2011, 286, 34426-34439, 10.1074/jbc.m111.257444.
- Charles Géminard; Aude De Gassart; Lionel Blanc; Michel Vidal; Degradation of AP2 During Reticulocyte Maturation Enhances Binding of Hsc70 and Alix to a Common Site on TfR for Sorting into Exosomes. Traffic 2004, 5, 181-193, 10.1111/j.1600-0854.2004.0167.x.
- Clotilde Théry; Muriel Boussac; Philippe Véron; Paola Ricciardi-Castagnoli; Graça Raposo; Jerôme Garin; Sebastian Amigorena; Proteomic Analysis of Dendritic Cell-Derived Exosomes: A Secreted Subcellular Compartment Distinct from Apoptotic Vesicles. The Journal of Immunology 2001, 166, 7309-7318, 10.4049/jimmunol.166.12.7309.
- Uwe Irion; Daniel St Johnston; bicoid RNA localization requires specific binding of an endosomal sorting complex. Nature 2007, 445, 554-558, 10.1038/nature05503.
- Ariel Savina; Claudio M. Fader; María T. Damiani; María Isabel Colombo; Rab11 Promotes Docking and Fusion of Multivesicular Bodies in a Calcium-Dependent Manner. Traffic 2004, 6, 131-143, 10.1111/j.1600-0854.2004.00257.x.
- Graça Raposo; Danielle Tenza; Salahedine Mecheri; Roger Peronet; Christian Bonnerot; Catherine Desaymard; Accumulation of Major Histocompatibility Complex Class II Molecules in Mast Cell Secretory Granules and Their Release upon Degranulation. Molecular Biology of the Cell 1997, 8, 2631-2645, 10.1091/mbc.8.12.2631.
- J. Fauré; G. Lachenal; M. Court; J. Hirrlinger; C. Chatellard-Causse; B. Blot; J. Grange; G. Schoehn; Y. Goldberg; V. Boyer; et al. Exosomes are released by cultured cortical neurones. Molecular and Cellular Neuroscience 2006, 31, 642-648, 10.1016/j.mcn.2005.12.003.
- Matias Ostrowski; Nuno B. Carmo; Sophie Krumeich; Isabelle Fanget; Graça Raposo; Ariel Savina; Catarina F. Moita; Kristine Schauer; Alistair N. Hume; Rui P. Freitas; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nature 2009, 12, 19-30, 10.1038/ncb2000.
- Dimitry A. Chistiakov; Alexander N. Orekhov; Yuri V. Bobryshev; Cardiac Extracellular Vesicles in Normal and Infarcted Heart. International Journal of Molecular Sciences 2016, 17, 63, 10.3390/ijms17010063.
- Anna Janowska-Wieczorek; Marcin Wysoczynski; Jacek Kijowski; Leah Marquez-Curtis; Bogdan Machalinski; Janina Ratajczak; Mariusz Z. Ratajczak; Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. International Journal of Cancer 2004, 113, 752-760, 10.1002/ijc.20657.
- Tae Hoon Lee; Esterina D’Asti; Nathalie Magnus; Khalid Al-Nedawi; Brian Meehan; Janusz Rak; Microvesicles as mediators of intercellular communication in cancer—the emerging science of cellular ‘debris’. Seminars in Immunopathology 2011, 33, 455-467, 10.1007/s00281-011-0250-3.
- Janusz Rak; Microparticles in Cancer. Seminars in Thrombosis and Hemostasis 2010, 36, 888-906, 10.1055/s-0030-1267043.
- Joshua L. Hood; Roman Susana San; Samuel A. Wickline; Exosomes Released by Melanoma Cells Prepare Sentinel Lymph Nodes for Tumor Metastasis. Cancer Research 2011, 71, 3792-3801, 10.1158/0008-5472.can-10-4455.
- Raghu Kalluri; Valerie S. LeBleu; The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977, 10.1126/science.aau6977.
- Sangiliyandi Gurunathan; Min-Hee Kang; Muniyandi Jeyaraj; Muhammad Qasim; Jin-Hoi Kim; Review of the Isolation, Characterization, Biological Function, and Multifarious Therapeutic Approaches of Exosomes. Cells 2019, 8, 307, 10.3390/cells8040307.
- Lin Zhang; Dihua Yu; Exosomes in cancer development, metastasis, and immunity. Biochimica et Biophysica Acta (BBA) - Bioenergetics 2019, 1871, 455-468, 10.1016/j.bbcan.2019.04.004.
- Sanchita Bhatnagar; Kazuhiko Shinagawa; Francis J. Castellino; Jeffrey S. Schorey; Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo. Blood 2007, 110, 3234-3244, 10.1182/blood-2007-03-079152.
- Pramod K. Giri; Nicole A. Kruh; Karen M. Dobos; Jeff S. Schorey; Proteomic analysis identifies highly antigenic proteins in exosomes from M. tuberculosis -infected and culture filtrate protein-treated macrophages. PROTEOMICS 2010, 10, 3190-3202, 10.1002/pmic.200900840.
- Jody Winter; Darren Letley; Joanne Rhead; John Atherton; Karen Robinson; Helicobacter pylori Membrane Vesicles Stimulate Innate Pro- and Anti-Inflammatory Responses and Induce Apoptosis in Jurkat T Cells. Infection and Immunity 2014, 82, 1372-1381, 10.1128/iai.01443-13.
- Jaswant K. Jutley; Jerry Kelleher; Peter Whelan; John Mikel; Cytosolic retinoic acid-binding protein in human prostatic dysplasia and neoplasia. The Prostate 1986, 11, 127-132, 10.1002/pros.2990110204.
- Wandy L. Beatty; Heinz-Joachim Ullrich; David G. Russell; Mycobacterial surface moieties are released from infected macrophages by a constitutive exocytic event. European Journal of Cell Biology 2001, 80, 31-40, 10.1078/0171-9335-00131.
- Wandy L. Beatty; David G. Russell; Identification of Mycobacterial Surface Proteins Released into Subcellular Compartments of Infected Macrophages. Infection and Immunity 2000, 68, 6997-7002, 10.1128/iai.68.12.6997-7002.2000.
- Clotilde Théry; Matias Ostrowski; Elodie Segura; Membrane vesicles as conveyors of immune responses. Nature Reviews Immunology 2009, 9, 581-593, 10.1038/nri2567.
- Angélique Bobrie; Marina Colombo; Graça Raposo; Clotilde Théry; Exosome Secretion: Molecular Mechanisms and Roles in Immune Responses. Traffic 2011, 12, 1659-1668, 10.1111/j.1600-0854.2011.01225.x.
- Sanchita Bhatnagar; Jeffrey S. Schorey; Exosomes Released from Infected Macrophages Contain Mycobacterium avium Glycopeptidolipids and Are Proinflammatory. Journal of Biological Chemistry 2007, 282, 25779-25789, 10.1074/jbc.m702277200.
- Jian-Jun Wang; Cai Chen; Ping-Fang Xie; Yi Pan; Yun-Hong Tan; Li-Jun Tang; Proteomic analysis and immune properties of exosomes released by macrophages infected with Mycobacterium avium. Microbes and Infection 2014, 16, 283-291, 10.1016/j.micinf.2013.12.001.
- Paras K. Anand; Ellis Anand; Christopher K. E. Bleck; Elsa Anes; Gareth Griffiths; Exosomal Hsp70 Induces a Pro-Inflammatory Response to Foreign Particles Including Mycobacteria. PLOS ONE 2010, 5, e10136, 10.1371/journal.pone.0010136.
- Jeffrey S. Schorey; Sanchita Bhatnagar; Exosome Function: From Tumor Immunology to Pathogen Biology. Traffic 2008, 9, 871-881, 10.1111/j.1600-0854.2008.00734.x.
- Prachi P. Singh; Victoria L. Smith; Petros C. Karakousis; Jeffery S. Schorey; Exosomes Isolated from Mycobacteria-Infected Mice or Cultured Macrophages Can Recruit and Activate Immune Cells In Vitro and In Vivo. The Journal of Immunology 2012, 189, 777-785, 10.4049/jimmunol.1103638.
- Prachi P. Singh; Christopher Lemaire; John C. Tan; Erliang Zeng; Jeffery S. Schorey; Exosomes Released from M.tuberculosis Infected Cells Can Suppress IFN-γ Mediated Activation of Naïve Macrophages. PLOS ONE 2011, 6, e18564, 10.1371/journal.pone.0018564.
- Kithiganahalli N. Balaji; Girija Goyal; Yeddula Narayana; Madduri Srinivas; Rashmi Chaturvedi; Saleemulla Mohammad; Apoptosis triggered by Rv1818c, a PE family gene from Mycobacterium tuberculosis is regulated by mitochondrial intermediates in T cells. Microbes and Infection 2007, 9, 271-281, 10.1016/j.micinf.2006.11.013.
- Pramod K. Giri; Jeffrey S. Schorey; Exosomes Derived from M. Bovis BCG Infected Macrophages Activate Antigen-Specific CD4+ and CD8+ T Cells In Vitro and In Vivo. PLOS ONE 2008, 3, e2461, 10.1371/journal.pone.0002461.
- Chenjie Yang; Geetha Chalasani; Yue-Harn Ng; Paul D. Robbins; Exosomes Released from Mycoplasma Infected Tumor Cells Activate Inhibitory B Cells. PLOS ONE 2012, 7, e36138, 10.1371/journal.pone.0036138.
- Laurence Abrami; Lucia Brandi; Mahtab Moayeri; Michael J. Brown; Bryan A. Krantz; Stephen H. Leppla; F. Gisou Van Der Goot; Hijacking Multivesicular Bodies Enables Long-Term and Exosome-Mediated Long-Distance Action of Anthrax Toxin. Cell Reports 2013, 5, 986-996, 10.1016/j.celrep.2013.10.019.
- Camille Ettelaie; Mary E.W. Collier; Nicola J. James; Chao Li; Induction of tissue factor expression and release as microparticles in ECV304 cell line by Chlamydia pneumoniae infection. Atherosclerosis 2007, 190, 343-351, 10.1016/j.atherosclerosis.2006.04.005.
- Kyla Frohlich; Ziyu Hua; Jin Wang; Li Shen; Isolation of Chlamydia trachomatis and membrane vesicles derived from host and bacteria. Journal of Microbiological Methods 2012, 91, 222-230, 10.1016/j.mimet.2012.08.012.
- S. N. Chatterjee; J. Das; Electron Microscopic Observations on the Excretion of Cell-wall Material by Vibrio cholerae. Journal of General Microbiology 1967, 49, 1-11, 10.1099/00221287-49-1-1.
- I. W. DeVoe; J. E. Gilchrist; Michael M. Frank; Joseph May; Thelma Gaither; Leonard Ellman; RELEASE OF ENDOTOXIN IN THE FORM OF CELL WALL BLEBS DURING IN VITRO GROWTH OF NEISSERIA MENINGITIDIS. Journal of Experimental Medicine 1973, 138, 1156-1167, 10.1084/jem.138.5.1156.
- Sarah R. Schooling; Terry J. Beveridge; Membrane Vesicles: an Overlooked Component of the Matrices of Biofilms. Journal of Bacteriology 2006, 188, 5945-5957, 10.1128/jb.00257-06.
- Marika Renelli; Valério Matias; Reggie Y. Lo; Terry J. Beveridge; DNA-containing membrane vesicles of Pseudomonas aeruginosa PAO1 and their genetic transformation potential. Microbiology 2004, 150, 2161-2169, 10.1099/mic.0.26841-0.
- David W. Dorward; Claude F. Garon; DNA Is Packaged within Membrane-Derived Vesicles of Gram-Negative but Not Gram-Positive Bacteria.. Applied and Environmental Microbiology 1990, 56, 1960-2, .
- Eloise J. O'Donoghue; Anne Marie Krachler; Mechanisms of outer membrane vesicle entry into host cells. Cellular Microbiology 2016, 18, 1508-1517, 10.1111/cmi.12655.
- Petr Broz; Denise M. Monack; Newly described pattern recognition receptors team up against intracellular pathogens. Nature Reviews Immunology 2013, 13, 551-565, 10.1038/nri3479.
- Rishi D. Pathirana; Maria Kaparakis-Liaskos; Bacterial membrane vesicles: Biogenesis, immune regulation and pathogenesis. Cellular Microbiology 2016, 18, 1518-1524, 10.1111/cmi.12658.
- Masahito Hashimoto; Taichi Matsumoto; Miwa Tamura-Nakano; Mami Ozono; Shuhei Hashiguchi; Yasuo Suda; Characterization of outer membrane vesicles of Acetobacter pasteurianus NBRC3283. Journal of Bioscience and Bioengineering 2018, 125, 425-431, 10.1016/j.jbiosc.2017.11.006.
- María-Alexandra Cañas; María-José Fábrega; Rosa Giménez; Josefa Badia; Laura Baldomà; Outer Membrane Vesicles From Probiotic and Commensal Escherichia coli Activate NOD1-Mediated Immune Responses in Intestinal Epithelial Cells. Frontiers in Microbiology 2018, 9, 498, 10.3389/fmicb.2018.00498.
- Pankaj Deo; Seong H. Chow; Iain D. Hay; Oded Kleifeld; Adam Costin; Kirstin D. Elgass; Jhih-Hang Jiang; Georg Ramm; Kipros Gabriel; Gordon Dougan; et al. Outer membrane vesicles from Neisseria gonorrhoeae target PorB to mitochondria and induce apoptosis. PLOS Pathogens 2018, 14, e1006945, 10.1371/journal.ppat.1006945.
- Lisa Kunsmann; Christian Rueter; Andreas Bauwens; Lilo Greune; Malte Glueder; Bjoern Kemper; Angelika Fruth; Sun Nyunt Wai; Xiaohua He; Roland Lloubes; et al. Virulence from vesicles: Novel mechanisms of host cell injury by Escherichia coli O104:H4 outbreak strain. Scientific Reports 2015, 5, 13252, 10.1038/srep13252.
- Johanna Rivera; Radames J. B. Cordero; Antonio S. Nakouzi; Susana Frases; André Nicola; Arturo Casadevall; Bacillus anthracis produces membrane-derived vesicles containing biologically active toxins. Proceedings of the National Academy of Sciences 2010, 107, 19002-19007, 10.1073/pnas.1008843107.
- Jon M. Davis; Humberto M. Carvalho; Susan B. Rasmussen; Alison D. O'brien; Cytotoxic Necrotizing Factor Type 1 Delivered by Outer Membrane Vesicles of Uropathogenic Escherichia coli Attenuates Polymorphonuclear Leukocyte Antimicrobial Activity and Chemotaxis. Infection and Immunity 2006, 74, 4401-4408, 10.1128/iai.00637-06.
- Jennifer M. Bomberger; Siying Ye; Daniel P. MacEachran; Katja Koeppen; Roxanna L. Barnaby; George A. O'toole; Bruce A. Stanton; A Pseudomonas aeruginosa Toxin that Hijacks the Host Ubiquitin Proteolytic System. PLOS Pathogens 2011, 7, e1001325, 10.1371/journal.ppat.1001325.
- Viveka Schaar; Stefan P. W. De Vries; Maria Laura A. Perez Vidakovics; Hester J. Bootsma; Lennart Larsson; Peter W. M. Hermans; Anders Bjartell; Matthias Mörgelin; Kristian Riesbeck; Multicomponent Moraxella catarrhalis outer membrane vesicles induce an inflammatory response and are internalized by human epithelial cells. Cellular Microbiology 2010, 13, 432-449, 10.1111/j.1462-5822.2010.01546.x.
- Maria Laura A. Perez Vidakovics; Johan Jendholm; Matthias Mörgelin; Anne Månsson; Christer Larsson; Lars-Olaf Cardell; Kristian Riesbeck; B Cell Activation by Outer Membrane Vesicles—A Novel Virulence Mechanism. PLOS Pathogens 2010, 6, e1000724, 10.1371/journal.ppat.1000724.
- Cherie Blenkiron; Denis Simonov; Anita Muthukaruppan; Peter Tsai; Priscila Dauros; Sasha Green; Jiwon Hong; Cristin G. Print; Simon Swift; Anthony R. Phillips; et al. Uropathogenic Escherichia coli Releases Extracellular Vesicles That Are Associated with RNA. PLOS ONE 2016, 11, e0160440, 10.1371/journal.pone.0160440.
- Katja Koeppen; Thomas H. Hampton; Michael Jarek; Maren Scharfe; Scott A. Gerber; Daniel W. Mielcarz; Elora G. Demers; Emily L. Dolben; John H. Hammond; Deborah A. Hogan; et al. A Novel Mechanism of Host-Pathogen Interaction through sRNA in Bacterial Outer Membrane Vesicles. PLOS Pathogens 2016, 12, e1005672, 10.1371/journal.ppat.1005672.
- Joeri Tulkens; Olivier De Wever; An Hendrix; Analyzing bacterial extracellular vesicles in human body fluids by orthogonal biophysical separation and biochemical characterization. Nature Protocols 2019, 15, 40-67, 10.1038/s41596-019-0236-5.
- Joeri Tulkens; Glenn Vergauwen; Jan Van Deun; Edward Geeurickx; Bert Dhondt; Lien Lippens; Marie-Angélique De Scheerder; Ilkka Miinalainen; Pekka Rappu; Bruno G De Geest; et al. Increased levels of systemic LPS-positive bacterial extracellular vesicles in patients with intestinal barrier dysfunction. Gut 2018, 69, 191-193, 10.1136/gutjnl-2018-317726.
- Régis Stentz; Ana L. Carvalho; Emily J. Jones; Simon R. Carding; Fantastic voyage: the journey of intestinal microbiota-derived microvesicles through the body. Biochemical Society Transactions 2018, 46, 1021-1027, 10.1042/bst20180114.
- Jin-Young Park; Juli Choi; YunJin Lee; Jung-Eun Lee; Eun-Hwa Lee; Hye-Jin Kwon; Jinho Yang; Bo-Ri Jeong; Yoon-Keun Kim; Pyung-Lim Han; et al. Metagenome Analysis of Bodily Microbiota in a Mouse Model of Alzheimer Disease Using Bacteria-derived Membrane Vesicles in Blood. Experimental Neurobiology 2017, 26, 369-379, 10.5607/en.2017.26.6.369.
- Gregory D. Poore; Evguenia Kopylova; Qiyun Zhu; Carolina Carpenter; Serena Fraraccio; Stephen Wandro; Tomasz Kosciolek; Stefan Janssen; Jessica Metcalf; Se Jin Song; et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 2020, 579, 567-574, 10.1038/s41586-020-2095-1.
- Asako Shimoda; Koji Ueda; Shin Nishiumi; Naoko Murata-Kamiya; Sada-Atsu Mukai; Shin-Ichi Sawada; Takeshi Azuma; Masanori Hatakeyama; Kazunari Akiyoshi; Exosomes as nanocarriers for systemic delivery of the Helicobacter pylori virulence factor CagA. Scientific Reports 2016, 6, 18346, 10.1038/srep18346.
- Ying Che; Biao Geng; Yue Xu; Xin Miao; Ling Chen; Xianmin Mu; Jinshun Pan; Chen Zhang; Ting Zhao; Chao Wang; et al. Helicobacter pylori -induced exosomal MET educates tumour-associated macrophages to promote gastric cancer progression. Journal of Cellular and Molecular Medicine 2018, 22, 5708-5719, 10.1111/jcmm.13847.
- Stefan Rose-John; The Soluble Interleukin 6 Receptor: Advanced Therapeutic Options in Inflammation. Clinical Pharmacology & Therapeutics 2017, 102, 591-598, 10.1002/cpt.782.
- John A. Lust; Kathleen A. Donovan; Michael P. Kline; Philip R. Greipp; Robert A. Kyle; Nita J. Maihle; Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine 1992, 4, 96-100, 10.1016/1043-4666(92)90043-q.
- Neele Schumacher; Dörte Meyer; Andre Mauermann; Jan von der Heyde; Janina Wolf; Jeanette Schwarz; Katharina Knittler; Gillian Murphy; Matthias Michalek; Christoph Garbers; et al. Shedding of Endogenous Interleukin-6 Receptor (IL-6R) Is Governed by A Disintegrin and Metalloproteinase (ADAM) Proteases while a Full-length IL-6R Isoform Localizes to Circulating Microvesicles. Journal of Biological Chemistry 2015, 290, 26059-26071, 10.1074/jbc.m115.649509.
- Yufan Chen; Xinqiong Wang; Y. Yu; Yuan Xiao; Jiebin Huang; Zhanyong Yao; Xuehua Chen; Tong Zhou; P. Li; Chundi Xu; et al. Serum exosomes of chronic gastritis patients infected withHelicobacter pylorimediate IL-1α expression via IL-6 trans-signalling in gastric epithelial cells. Clinical & Experimental Immunology 2018, 194, 339-349, 10.1111/cei.13200.
- Jianjun Wang; Zhiyong Deng; Zeyou Wang; Jianhong Wu; Tao Gu; Yibiao Jiang; Guangxin Li; MicroRNA-155 in exosomes secreted from helicobacter pylori infection macrophages immunomodulates inflammatory response. American journal of translational research 2016, 8, 3700-3709, .
- Hyun-Il Choi; Jun-Pyo Choi; Jiwon Seo; Beom Jin Kim; Mina Rho; Jin Kwan Han; Jae Gyu Kim; Helicobacter pylori-derived extracellular vesicles increased in the gastric juices of gastric adenocarcinoma patients and induced inflammation mainly via specific targeting of gastric epithelial cells. Experimental & Molecular Medicine 2017, 49, e330-e330, 10.1038/emm.2017.47.
- Yalda Khosravi; Yakhya Dieye; Bee Hoon Poh; Chow Goon Ng; Mun Fai Loke; Khean Lee Goh; Jamuna Vadivelu; Culturable Bacterial Microbiota of the Stomach ofHelicobacter pyloriPositive and Negative Gastric Disease Patients. The Scientific World Journal 2014, 2014, 1-10, 10.1155/2014/610421.
- Denisse Bravo; Anilei Hoare; Cristopher Soto; Manuel A Valenzuela; Andrew Fg Quest; Helicobacter pyloriin human health and disease: Mechanisms for local gastric and systemic effects. World Journal of Gastroenterology 2018, 24, 3071-3089, 10.3748/wjg.v24.i28.3071.
- Hossein Ajdarkosh; Fahimeh Safarnezhad Tameshkel; Masoud Reza Sohrabi; Mohammad Hadi Karbalaie Niya; Gholamreza Hemmasi; Bahare Amirkalali; Farhad Zamani; Ameneh Mehraban Sarasiab; Association of Helicobacter Pylori Infection with Colon Polyp and Colorectal Cancer. British Journal of Medicine and Medical Research 2016, 16, 1-6, 10.9734/bjmmr/2016/26480.
- Harvey A. Risch; Lingeng Lu; Mark S. Kidd; Jing Wang; Wei Zhang; Quanxing Ni; Yu-Tang Gao; Herbert Yu; Helicobacter pylori Seropositivities and Risk of Pancreatic Carcinoma. Cancer Epidemiology Biomarkers & Prevention 2013, 23, 172-178, 10.1158/1055-9965.epi-13-0447.
- Aaron T. Irving; Hitomi Mimuro; Thomas A. Kufer; Camden Lo; Richard Wheeler; Lorinda J. Turner; Belinda J. Thomas; Christian Malosse; Michael P. Gantier; Linda N. Casillas; et al. The Immune Receptor NOD1 and Kinase RIP2 Interact with Bacterial Peptidoglycan on Early Endosomes to Promote Autophagy and Inflammatory Signaling. Cell Host & Microbe 2014, 15, 623-635, 10.1016/j.chom.2014.04.001.
- Lorinda Turner; Natalie J. Bitto; David L. Steer; Camden Lo; Kimberley D’Costa; Georg Ramm; Mitch Shambrook; Andrew F. Hill; Richard L. Ferrero; Maria Kaparakis-Liaskos; et al. Helicobacter pylori Outer Membrane Vesicle Size Determines Their Mechanisms of Host Cell Entry and Protein Content. Frontiers in Immunology 2018, 9, 1466, 10.3389/fimmu.2018.01466.
- Lauren Zavan; Natalie J Bitto; Ella L. Johnston; David W. Greening; Maria Kaparakis‐Liaskos; Helicobacter pyloriGrowth Stage Determines the Size, Protein Composition, and Preferential Cargo Packaging of Outer Membrane Vesicles. PROTEOMICS 2018, 19, e1800209, 10.1002/pmic.201800209.
- Roberto Fiocca; Vittorio Necchi; Patrizia Sommi; Vittorio Ricci; John Telford; Timothy L. Cover; Enrico Solcia; Release ofHelicobacter pylori vacuolating cytotoxin by both a specific secretion pathway and budding of outer membrane vesicles. Uptake of released toxin and vesicles by gastric epithelium. The Journal of Pathology 1999, 188, 220-226, 10.1002/(sici)1096-9896(199906)188:2<220::aid-path307>3.0.co;2-c.
- Jacqueline Keenan; Tony Day; Stephanie Neal; Bramwell Cook; Guillermo Perez-Perez; Randall Allardyce; Philip Bagshaw; A role for the bacterial outer membrane in the pathogenesis of Helicobacter pylori infection. FEMS Microbiology Letters 2000, 182, 259-264, 10.1016/s0378-1097(99)00592-3.
- Ursula Heczko; Valerie C Smith; R Mark Meloche; Alison M.J Buchan; B.Brett Finlay; Characteristics of Helicobacter pylori attachmentto human primary antral epithelial cells. Microbes and Infection 2000, 2, 1669-1676, 10.1016/s1286-4579(00)01322-8.
- Kenny Chitcholtan; Mark B. Hampton; Jacqueline I. Keenan; Outer membrane vesicles enhance the carcinogenic potential of Helicobacter pylori. Carcinogenesis 2008, 29, 2400-2405, 10.1093/carcin/bgn218.
- Erica Mullaney; Paul A. Brown; Sinead M. Smith; Catherine H. Botting; Yoshio Y. Yamaoka; Ana M. Terres; Dermot P. Kelleher; Henry J. Windle; Proteomic and functional characterization of the outer membrane vesicles from the gastric pathogen Helicobacter pylori. PROTEOMICS – Clinical Applications 2009, 3, 785-796, 10.1002/prca.200800192.
- Annelie Olofsson; Anna Vallström; Katja Petzold; Nicole Tegtmeyer; Jürgen Schleucher; Sven Carlsson; Rainer Haas; Steffen Backert; Sun Nyunt Wai; Gerhard Gröbner; et al. Biochemical and functional characterization of Helicobacter pylori vesicles. Molecular Microbiology 2010, 77, 1539-1555, 10.1111/j.1365-2958.2010.07307.x.
- J A Collett; M J Burt; C M Frampton; K H Yeo; T M Chapman; R C Buttimore; H B Cook; B A Chapman; Seroprevalence of Helicobacter pylori in the adult population of Christchurch: risk factors and relationship to dyspeptic symptoms and iron studies.. The New Zealand medical journal 1999, 112, 292–295, .
- R. Yip; Pervasive occult gastrointestinal bleeding in an Alaska native population with prevalent iron deficiency. Role of Helicobacter pylori gastritis. JAMA 1997, 277, 1135-1139, 10.1001/jama.277.14.1135.
- ‡ Nils Milman; Steffen Rosenstock‡; Leif Andersen§; Torben Jørgensen‡; Olaf Bonnevie∥; Serum ferritin, hemoglobin, and Helicobacter pylori infection: A seroepidemiologic survey comprising 2794 Danish adults. Gastroenterology 1998, 115, 268-274, 10.1016/s0016-5085(98)70192-1.
- Jacqueline I. Keenan; Kylie A. Davis; Clare R. Beaugie; Joseph J. McGovern; Anthony P. Moran; Alterations in Helicobacter pylori outer membrane and outer membrane vesicle-associated lipopolysaccharides under iron-limiting growth conditions*. Innate Immunity 2008, 14, 279-290, 10.1177/1753425908096857.
- Jacqueline I. Keenan; Randall A. Allardyce; Iron influences the expression of Helicobacter pylori outer membrane vesicle-associated virulence factors. European Journal of Gastroenterology & Hepatology 2000, 12, 1267-1273, 10.1097/00042737-200012120-00002.
- Sean O. Hynes; Jacqueline I. Keenan; John A. Ferris; Heidi Annuk; Anthony P. Moran; Lewis Epitopes on Outer Membrane Vesicles of Relevance to Helicobacter pylori Pathogenesis. Helicobacter 2005, 10, 146-156, 10.1111/j.1523-5378.2005.00302.x.
- Annelie Olofsson; Lars Nygård Skalman; Ikenna Obi; Richard Lundmark; Anna Arnqvist; Uptake of Helicobacter pylori Vesicles Is Facilitated by Clathrin-Dependent and Clathrin-Independent Endocytic Pathways. mBio 2014, 5, e00979-14-14, 10.1128/mbio.00979-14.
- Heather Parker; Kenny Chitcholtan; Mark B. Hampton; Jacqueline I. Keenan; Uptake of Helicobacter pylori Outer Membrane Vesicles by Gastric Epithelial Cells. Infection and Immunity 2010, 78, 5054-5061, 10.1128/iai.00299-10.
- Jacqueline I Keenan; Randall A Allardyce; Philip F Bagshaw; Dual silver staining to characterise Helicobacter spp. outer membrane components. Journal of Immunological Methods 1997, 209, 17-24, 10.1016/s0022-1759(97)00141-5.
- Vittorio Ricci; Valentina Chiozzi; Vittorio Necchi; Amanda Oldani; Marco Romano; Enrico Solcia; Ulderico Ventura; Free-soluble and outer membrane vesicle-associated VacA from Helicobacter pylori: Two forms of release, a different activity. Biochemical and Biophysical Research Communications 2005, 337, 173-178, 10.1016/j.bbrc.2005.09.035.
- Jung Hwa Lee; So Hyun Jun; Jung-Min Kim; Seung Chul Baik; Je Chul Lee; Morphological changes in human gastric epithelial cells induced by nuclear targeting of Helicobacter pylori urease subunit A. Journal of Microbiology 2015, 53, 406-414, 10.1007/s12275-015-5085-5.
- Manuel Valenzuela-Valderrama; Paulina Cerda-Opazo; Steffen Backert; María Fernanda González; Nicolás Carrasco-Véliz; Carla Jorquera-Cordero; Sergio Wehinger; Jimena Canales; Denisse Bravo; Andrew F. G. Quest; et al. The Helicobacter pylori Urease Virulence Factor Is Required for the Induction of Hypoxia-Induced Factor-1α in Gastric Cells. Cancers 2019, 11, 799, 10.3390/cancers11060799.
- Sujinna Lekmeechai; Yu-Ching Su; Marta Brant; Maria Alvarado-Kristensson; Anna Vallström; Ikenna Obi; Anna Arnqvist; Kristian Riesbeck; Helicobacter pylori Outer Membrane Vesicles Protect the Pathogen From Reactive Oxygen Species of the Respiratory Burst. Frontiers in Microbiology 2018, 9, 1837, 10.3389/fmicb.2018.01837.
- Hideo Yonezawa; Takako Osaki; Satoshi Kurata; Minoru Fukuda; Hayato Kawakami; Kuniyasu Ochiai; Tomoko Hanawa; Shigeru Kamiya; Outer Membrane Vesicles of Helicobacter pylori TK1402 are Involved in Biofilm Formation. BMC Microbiology 2008, 9, 197-197, 10.1186/1471-2180-9-197.
- Magdalena Chmiela; Natalia Walczak; Karolina Rudnicka; Helicobacter pylori outer membrane vesicles involvement in the infection development and Helicobacter pylori-related diseases. Journal of Biomedical Science 2018, 25, 78, 10.1186/s12929-018-0480-y.
- Maria V. Turkina; Annelie Olofsson; Karl-Eric Magnusson; Anna Arnqvist; Elena Vikström; Helicobacter pylori vesicles carrying CagA localize in the vicinity of cell–cell contacts and induce histone H1 binding to ATP in epithelial cells. FEMS Microbiology Letters 2015, 362, p. fnv076., 10.1093/femsle/fnv076.
- Salim Ismail; Mark B. Hampton; Jacqueline I. Keenan; Helicobacter pylori Outer Membrane Vesicles Modulate Proliferation and Interleukin-8 Production by Gastric Epithelial Cells. Infection and Immunity 2003, 71, 5670-5675, 10.1128/iai.71.10.5670-5675.2003.
- Guadalupe Ayala; Luz Torres; Magali Espinosa; Geny Fierros-Zarate; Vilma Maldonado; Jorge Melã©Ndez-Zajgla; External membrane vesicles from Helicobacter pylori induce apoptosis in gastric epithelial cells. FEMS Microbiology Letters 2006, 260, 178-185, 10.1111/j.1574-6968.2006.00305.x.
- Barry D. Hock; Judith L. McKenzie; Jacqueline I. Keenan; Helicobacter pylori outer membrane vesicles inhibit human T cell responses via induction of monocyte COX-2 expression. Pathogens and Disease 2017, 75, ftx034, 10.1093/femspd/ftx034.
- Jacqueline Keenan; Jane Oliaro; Neil Domigan; Howard Potter; Geoff Aitken; Randall Allardyce; Justin Roake; Immune Response to an 18-Kilodalton Outer Membrane Antigen Identifies Lipoprotein 20 as a Helicobacter pyloriVaccine Candidate. Infection and Immunity 2000, 68, 3337-3343, 10.1128/iai.68.6.3337-3343.2000.
- Francis F. Arhin; France Moreau; James W. Coulton; Elaine L. Mills; Outer Membrane Proteins and Serosubtyping with Outer Membrane Vesicles from Clinical Isolates of Neisseria meningitidis. Current Microbiology 1996, 34, 18-22, 10.1007/s002849900137.
- Samantha Flores-Treviño; Soraya Mendoza-Olazarán; Paola Bocanegra-Ibarias; Héctor Jesús Maldonado-Garza; Elvira Garza-González; Helicobacter pyloridrug resistance: therapy changes and challenges. Expert Review of Gastroenterology & Hepatology 2018, 12, 819-827, 10.1080/17474124.2018.1496017.
- Qiong Liu; Xiuzhen Li; Yingxuan Zhang; Zifan Song; Ruizhen Li; Huan Ruan; Xiaotian Huang; Orally-administered outer-membrane vesicles from Helicobacter pylori reduce H. pylori infection via Th2-biased immune responses in mice. Pathogens and Disease 2019, 77, ftz050, 10.1093/femspd/ftz050.
- Lorinda Turner; Judyta Praszkier; Melanie L. Hutton; David Steer; Georg Ramm; Maria Kaparakis-Liaskos; Richard L. Ferrero; Increased Outer Membrane Vesicle Formation in aHelicobacter pylori tolBMutant. Helicobacter 2015, 20, 269-283, 10.1111/hel.12196.
- Zifan Song; Biaoxian Li; Yingxuan Zhang; Ruizhen Li; Huan Ruan; Jing Wu; Qiong Liu; Outer Membrane Vesicles of Helicobacter pylori 7.13 as Adjuvants Promote Protective Efficacy Against Helicobacter pylori Infection. Frontiers in Microbiology 2020, 11, 1340, 10.3389/fmicb.2020.01340.
- Anthony Axon; Symptoms and diagnosis of gastric cancer at early curable stage. Best Practice & Research Clinical Gastroenterology 2006, 20, 697-708, 10.1016/j.bpg.2006.03.015.
- Aleksander Szymczak; Stanisław Ferenc; Joanna Majewska; Paulina Miernikiewicz; Jan Gnus; Wojciech Witkiewicz; Krystyna Dąbrowska; Application of 16S rRNA gene sequencing in Helicobacter pylori detection. PeerJ 2020, 8, e9099, 10.7717/peerj.9099.
- Gauri Godbole; Francis Mégraud; Emilie Bessède; Review: Diagnosis of Helicobacter pylori infection. Helicobacter 2020, 25, e12735, 10.1111/hel.12735.
- Takeshi Yasuda; Tomoyuki Hiroyasu; Satoru Hiwa; Yuto Okada; Sadanari Hayashi; Yuki Nakahata; Yuriko Yasuda; Tatsushi Omatsu; Akihiro Obora; Takao Kojima; et al. Potential of automatic diagnosis system with linked color imaging for diagnosis of Helicobacter pylori infection. Digestive Endoscopy 2020, 32, 373-381, 10.1111/den.13509.
- Ana Isabel Lopes; Filipa F Vale; Mónica Oleastro; Helicobacter pylori infection - recent developments in diagnosis. World Journal of Gastroenterology 2014, 20, 9299-9313, 10.3748/wjg.v20.i28.9299.
- Taher Mohammadian; Leila Ganji; The Diagnostic Tests for Detection ofHelicobacter pyloriInfection. Monoclonal Antibodies in Immunodiagnosis and Immunotherapy 2019, 38, 1-7, 10.1089/mab.2018.0032.
- J Sreekumar; N France; S Taylor; T Matthews; P Turner; P Bliss; Alan H Brook; Ajm Watson; Diagnosis of Helicobacter pylori by carbon-13 urea breath test using a portable mass spectrometer. SAGE Open Medicine 2015, 3, p. 2050312115569565., 10.1177/2050312115569565.
- Yao-Kuang Wang; Fu-Chen Kuo; Chung-Jung Liu; Meng-Chieh Wu; Hsiang-Yao Shih; Sophie Sw Wang; Jeng-Yih Wu; Chao-Hung Kuo; Yao-Kang Huang; Deng-Chyang Wu; et al. Diagnosis ofHelicobacter pyloriinfection: Current options and developments. World Journal of Gastroenterology 2014, 21, 11221-11235, 10.3748/wjg.v21.i40.11221.
- Julia Butt; William J. Blot; Martha J. Shrubsole; Matthew G. Varga; Laura H. Hendrix; Sydnee Crankshaw; Tim Waterboer; Michael Pawlita; Meira Epplein; Performance of multiplex serology in discriminating active vs past Helicobacter pylori infection in a primarily African American population in the southeastern United States. Helicobacter 2019, 25, e12671-e12671, 10.1111/hel.12671.
- Lucio Barile; Giuseppe Vassalli; Exosomes: Therapy delivery tools and biomarkers of diseases. Pharmacology & Therapeutics 2017, 174, 63-78, 10.1016/j.pharmthera.2017.02.020.
- Ellen Namork; Petter Brandtzaeg; Fatal meningococcal septicaemia with “blebbing” meningococcus. The Lancet 2002, 360, 1741, 10.1016/s0140-6736(02)11721-1.