Neurodegenerative disorders are desperately lacking treatment options. It is imperative that drug repurposing be considered in the fight against neurodegenerative diseases. Fenamates have been studied for efficacy in treating several neurodegenerative diseases.
- tolfenamic acid
- mefenamic acid
- flufenamic acid
- meclofenamic acid
- fenamate
- NSAID
- neurodegenerative
- Alzheimer’s disease
1. Introduction
2. Fenamate Pharmacokinetics and Pharmacodynamics
2.1. Tolfenamic Acid
2.2. Mefenamic Acid
2.3. Meclofenamic Acid
2.4. Flufenamic Acid
2.5. Adverse Drug Reactions
3. Fenamates and Alzheimer’s Disease (AD)
4. Fenamates and Cognitive Impairment
As healthcare has improved, life expectancy has extended, leading to substantial increases in the number of individuals over the age of 65 years old who are at risk of developing cognitive impairment and dementia [54]. Mild cognitive impairment (MCI), a term used for over four decades, describes individuals with a noticeable decline in cognitive abilities that does not interfere with their daily functioning [55]. There are many different etiologies of MCI, including vascular, neurodegenerative, psychiatric, and medical [56][57][58]. Pharmacological interventions are limited and systematic reviews using randomized controlled trials have not investigated a protective effect of dementia medications or NSAIDs [54]. The following section focuses on studies where fenamates have been used to treat non-AD-related cognitive impairment from different etiologies.
4.1. Fenamates and Tauopathies
In AD, the characteristic hallmarks are Aβ plaques and neurofibrillary tau tangles (NFTs), and the earliest symptom of AD is cognitive impairment, as mentioned above. Many studies have suggested that the progression of mild cognitive impairment found in AD is most closely associated with NFT formation than Aβ plaque load [43]. Tau aggregation is not unique to AD; in fact, tau aggregation accounts for more than 20 neurological disorders known as tauopathies, which include AD, frontotemporal dementia with parkinsonism-17 (FTD-17), progressive supranuclear palsy (PSP), corticobasal degeneration, chronic traumatic encephalopathy, and Parkinson’s disease (PD) [59]. Moreover, therapeutics that are useful for one tauopathy may be therapeutic in others to combat cognitive decline. In fact, TA has been designated by the FDA and European Medicines Agency (EMA) as an orphan drug for the treatment of PSP, with a clinical trial currently under final approval to evaluate TA in individuals with PSP (NCT04253132).
4.2. Fenamates and Chronic Alcohol Exposure
Alcoholism is a chronic disorder, accounting for 5.3% of all deaths worldwide, that is known to be highly correlated with multiple neuropsychiatric diseases and cognitive impairment [30][60]. Several studies have shown that over-consumption of alcohol is a risk factor for dementia via many proposed pathways. Chronic alcohol use and withdrawal may stimulate Toll-like receptors (TLRs) 2 and 4, which could directly lead to microglial activation, neuroinflammation, and neuronal death [30]. Studies have also demonstrated that alcoholic individuals have increased Iba-1 expression, which is a marker of microglial expression [61].
Rajesh et al. (2017) observed that chronic alcohol exposure produced cognitive impairment in zebrafish, evidenced by the inability to retain the memory of a learned task, whereas zebrafish exposed to alcohol and MFA significantly retained memory of the learned task [62]. Furthermore, MFA exposure decreased acetylcholine esterase (AChE) activity in the brain of alcohol-exposed zebrafish [62]. The proposed mechanism for the protective effect was a generation of MFA free radicals during interaction with peroxidase, which was originally reported by Muraoka and Miura (2009) [22].
4.3. Fenamates and Ischemic Injury
Cerebral ischemia due to cerebral vessel blockage is a leading cause of death worldwide and neurons are uniquely vulnerable to ischemic injury [63][64]. According to the American Heart Association, stroke is the fifth leading cause of death in the United States [65]. Stroke is typically classified into two categories: ischemic or hemorrhagic [66]. Glutamate-induced excitotoxicity, reperfusion injury due to reactive oxygen species (ROS), neuroinflammation mediated by excessive microglia activation, and impaired axonal regeneration have all been proposed as potential underlying mechanisms of ischemic brain injury [67].
Studies have shown that many fenamates have neuroprotective effects against excitotoxicity-induced cell death [64][68]. MFA and MCFA have been proven to be neuroprotective against glutamate-evoked excitotoxicity in cultured embryonic rat hippocampal neurons, and intracerebroventricular (ICV) administration of MFA for 24 hours reduced brain damage in rodents [68]. In another study, treatment with MFA reduced cerebral edema, infarct volume, total ischemic brain damage, and edema, which provided evidence that fenamate NSAIDs may be neuroprotective against ischemic stroke [69].
5. Fenamates and Huntington’s Disease
Huntington’s disease (HD) is an autosomal-dominant neurodegenerative disorder caused by the abnormal expansion of the cytosine–adenine–guanine (CAG) repeat in the IT15 gene located on chromosome 4 [70]. This results in the production of mutant huntingtin (mHtt) protein with a long polyglutamine stretch in the N-terminus region of the Huntingtin protein (Htt) [70][71]. Individuals with greater than 39 CAG repeats develop HD, while those with 36–39 have reduced penetrance [71].
The mean prevalence of HD is estimated at 5.5 cases per 100,000 in the world [72]. Common symptoms are motor dysfunction, psychiatric disturbance, and cognitive deficits including chorea and loss of coordination [71][73]. mHtt causes selective neuronal loss in the striatum (caudate nucleus and putamen) with specific loss of efferent medium spiny neurons (MSNs) and dysfunction in the brain through multiple mechanisms [71][73]. The presence of mutant huntingtin aggregates found initially in the nucleus and later in the cytoplasm and neuronal processes, are considered the hallmark of HD [73].
The most common mouse model of HD is the R6 transgenic model that expresses a truncated form of the human Htt and has been used to examine therapeutic strategies [70]. The human htt gene is among many that are regulated by Sp1, which suggests that TA may be an effective treatment to attenuate motor and cognitive deficits for HD patients [74]. In fact, R6/1 mice treated with TA demonstrated improved motor performance in the rotarod test and attenuated cognitive decline, as observed during the novel object recognition (NOR) and passive avoidance tests [70]. Furthermore, TA treatment significantly decreased protein expression of mHtt and SP1 in vivo, and alleviated oxidative stress in PC12 cells [70]. These results suggest that TA treatment may facilitate the clearance of mutant Htt aggregates by SP1 inhibition and activation of the autophagy pathway [70].
6. Fenamates and Epilepsy
Epilepsy is a neurological disorder characterized by recurrent unprovoked seizures and is the fourth most common neurological disorder affecting approximately 50-65 million people worldwide [75][76]. Cognitive impairment, mood, and behavior disorders are common comorbidities of epilepsy [77]. Epileptic dementia is a term that was coined in the 19th century based on the idea that epilepsy causes progressive cognitive decline [77]. Currently, epilepsy therapeutics target ion channels or neurotransmitter systems; however, these treatments only provide relief for 60% of patients [76]. The following section reviews how fenamates are known to modulate several ion channels that are implicated in epileptogenesis.
Physiologically, epilepsy is linked to neuronal hyperexcitability [78]. The regulation of excitability is partially controlled by subthreshold, voltage-gated K+ currents (M-currents) that are generated by M-type K+ channels [78]. The low-threshold gating and slow activation and deactivation of the M-current provide relief from repetitive firing and neuronal excitability [78][79]. According to Peretz et al. (2005), MCFA is a potent and specific opener of KCNQ2/Q3 channels, enhanced M-currents, and reduced evoked action potentials [78].
Voltage-gated sodium channels (VGSC) have also been linked to neuronal disorders such as epilepsy, autism, muscle disease, and pain [80]. The VGSC subtypes Nav1.7 and Nav1.8 have been implicated as targets for pain management [80]. MFA, FFA, and TA inhibited peak sodium currents and significantly affected the inactivation processes of hNav1.7 and hNav1.8 with I-V curves left-shifted to the hyperpolarized direction. These findings may contribute to the well-known analgesic effects of these fenamates [80]. In another study, FFA inhibited VGSC currents in hippocampal pyramidal neurons by slowing down the inactivation process of the sodium current and shifting the inactivation curve toward more hyperpolarized potentials [81].
7. Overview of Alternative Drugs
In this review, we have discussed three key neurodegenerative pathways modulated by fenamates (Figures 2 and 3). It is also imperative to acknowledge the originality of each major mechanism and other drugs that may provide similar effects. The following section overviews the currently approved or potentially therapeutic drugs that use similar molecular mechanisms as stated above.
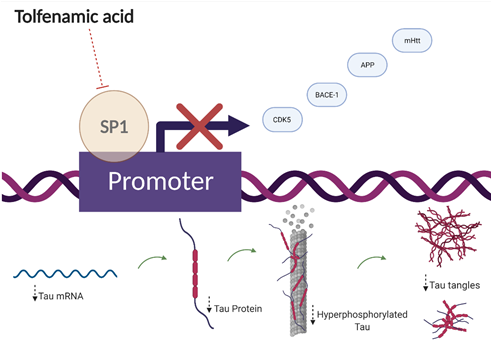
Figure 2. Proposed transcriptional mechanism of action for tolfenamic acid. Tolfenamic acid inhibits specificity protein 1 (SP1)-DNA binding, which leads to decreased expression of amyloid precursor protein (APP), mutant huntingtin protein (mHtt), β-secretase-1 (BACE-1), and cyclin-dependent kinase-5 (CDK5). The bottom half of the illustration shows the impact of tolfenamic acid on tau tangles via SP1 inhibition.
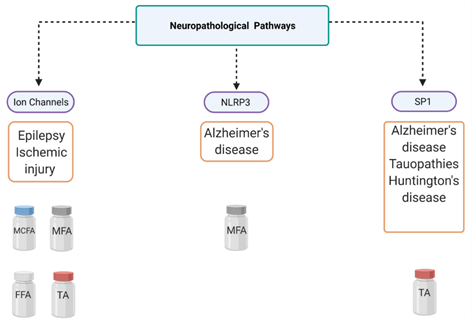
Figure 3. Mechanistic overview of fenamate neuropharmacology. This figure illustrates several diseases that can be targeted using fenamates and their three major pathways of interest. Tolfenamic acid (TA), mefenamic acid (MFA), meclofenamic acid (MCFA), flufenamic acid (FFA), NLR family pyrin domain-containing 3 (NLRP3), specificity protein 1 (SP1).
In the past, NSAIDs were extensively studied for their possible role as cancer therapeutics. Chronic inflammation has been linked widely to carcinogenesis, and epidemiological studies have indicated the chemoprotective and chemopreventive properties of NSAIDs [82]. Fenamate NSAIDs are known to modulate several pathways and exhibit anti-tumor activities in models for several cancers [83][84][85][86]. In fact, The SP1 hypothesis for AD intervention originated from cancer research as SP1 is upregulated in various types of cancer [87]. Although TA is unique among NSAIDs for its’ ability to target SP1, TA is not the only known therapeutic able to modulate SP1 binding and activity. Mithramycin (MTM) is an antineoplastic antibiotic that has been proven to bind to GC-rich DNA sequences and thus interferes with transcription factors that bind to GC-rich DNA regions, such as SP1 [87][88][89]. It has also been suggested that MTM can selectively inhibit SP1 but the mechanism is not well understood [87]. MTM treatment reduced cerebral Aβ levels and plaque burden, inhibited APP processing, alleviated tau hyperphosphorylation, and inhibited phosphorylated CDK5 and GSK3β pathways in APPswe/PS1dE9 mice [90]. MTM treatment also upregulated synaptic plasticity gene expression in an AD model in vitro, and prolonged survival and improved motor performance in an HD mouse model [89][91]. Collectively, these reports support the SP1 hypothesis for neurodegenerative disease intervention.
Furthermore, the role of COX inhibition in neurodegenerative and neuroinflammatory treatment or prevention is pertinent to mention. COX-2 is expressed under basal conditions in neuronal regions that overlap with AD pathogenesis [92]. It has even been suggested that the overexpression of neuronal COX-2 activity may disrupt normal neuronal function [92]. Due to the evidence linking COX-2 dysregulation to neurodegenerative diseases, NSAIDs have been implicated for their potential therapeutic effects in neurodegenerative disease states, particularly AD. In APP-PS1 mice, administration with ibuprofen during early adulthood prevented memory deficits [92]. However, the correlation of COX inhibition and the decreased occurrence or progression of neurodegenerative diseases has been debated because, although epidemiological data have shown that populations with long histories of NSAID use were at lower risk of AD, clinical studies have failed to achieve significant effects in the treatment of AD [93]. These findings may suggest the idea that NSAIDs may be useful in neurodegeneration prevention but are less useful for treatment; however, there has been no conclusive data proving their efficacy in prevention in humans.
Alzheimer’s disease is the only top-10 cause of death without a disease-modifying treatment approved by the FDA. Several recent anti-amyloid therapies have failed in clinical trials, which has prompted researchers to explore alternative pathways to target [94]. Targeting neuroinflammation is a recent approach that has gained more attention due to the high failure rate of previous drug candidates [94]. The NLRP3 inflammasome is a well-known driver of tau pathology and there are several proven direct and indirect inhibitors of the NLRP3 inflammasome that have been identified [48][95][96]. However, inhibition of VRAC by MFA is a novel approach to NLRP3 inhibition, and researchers have suggested that it is important to consider indirect therapeutic methods as targeting the inflammasome itself may result in peripheral complications [97].
Anti-seizure drugs (ASDs) are the predominant form of treatment for symptomatic relief for patients living with epilepsy [98]. ASDs interact with a variety of cellular targets including voltage-gated ion channels, GABAA receptors, GABA transporter 1 (GAT), synaptic vesicle proteins, ionotropic glutamate receptors, and several other targets of interest [98]. Currently, there are about 30 ASDs on the market [98]. While targeting VGSCs is not a novel mechanism, to our knowledge, there are no current commonly used drugs that target KCNQ2/Q3 channels [99]. Retigabine was discovered in the 1980s as a potent opener of channels formed by KCNQ2 and KCNQ3 subunits and was used to treat neonatal epileptic encephalopathy and, unfortunately, it was discontinued in 2017 due to pigmentary changes induced in skin, mucosae, and eyes [79][100]. For acute ischemic stroke, glutamate-induced excitotoxicity is a well-known contributor to ischemic neuronal death and has been targeted indirectly by NMDA receptor antagonists [101]. Thus far, there are no FDA-approved drugs for cerebral ischemia that target glutamate excitotoxicity, to our knowledge, as several have failed in randomized controlled clinical trials in humans [101]. There have been recent novel advances to combat glutamate-excitotoxicity, such as introducing glutamate scavengers or “grabbers”, which further reiterates the idea that glutamate excitotoxicity remains an important target for cerebral ischemia [102].
8. Discussion and Conclusions
Altogether, we have presented mounting evidence of several non-canonical disease pathways, from Alzheimer’s disease to epilepsy, that are modulated by various fenamates (Figures 2 and 3), and there are several significant conclusions to be mentioned in terms of this review. First, it is important to consider fenamates in neuropathological drug development as modulating several pathways may provide greater protective effects than molecules that have single targets, which may allow them to be efficacious at lower doses [2]. The main benefit may be in lowering inflammation as well as other disease-specific targets. Fenamates seem to act directly on their targets such as enzymes and ion channels, as well as indirectly by modulating transcription factors and thus impacting disease-specific gene expression. Interestingly, the ability of certain fenamates to activate proteasome-dependent degradation of SP1, and inhibit NLRP3 and certain ion channels is unique among NSAIDs, which may be indicative of a chemical class effect instead of a therapeutic class effect (NSAID) [2][34][103]. Drug repurposing offers many benefits as it may expedite the drug discovery process by shortening the high-throughput screening and clinical trial phases and provide proven safe alternatives to existing treatments. This is particularly important as CNS drugs have high failure rates and tend to have poorly understood pathways, and often patients have several co-morbidities. Furthermore, it is important to consider the adverse reactions that have been well documented due to long-term NSAID use and how the length of therapy and the therapeutic dose required may determine the safety of these small molecules for age-related disorders. In fact, fenamates could serve as a scaffold in future therapeutic drug design for neurodegenerative diseases. Finally, these data suggest the effect that fenamates have is polyvalent and substantiates the hypothesis that some therapeutic effects are independent of the COX pathway. In the future, exploratory pathway analysis and a comprehensive safety profile for each drug could provide insight into drug mechanisms of action and possible adverse effects specific to fenamates.
Abbreviations
Nonsteroidal anti-inflammatory drugs (NSAIDS)
Cyclooxygenases (COX)
Prostaglandin (PG)
Inhibitory concentration (IC50)
Mefenamic acid (MFA)
Tolfenamic acid (TA)
Meclofenamic acid (MCFA)
Flufenamic acid (FFA)
Prostaglandin H synthase (PHGS)
Alzheimer’s disease (AD)
Magnetic resonance imaging (MRI)
Cerebral spinal fluid (CSF)
Late-onset Alzheimer’s disease (LOAD)
Early-onset Alzheimer’s disease (EOAD)
Federal Drug Administration (FDA)
N-Methyl-D-aspartic acid (NMDA)
Amyloid precursor protein (APP)
β-Secretase-1 (BACE-1)
Specificity protein 1 (SP1)
Morris water maze (MWM)
Microtubule-associated protein tau (MAPT)
Cyclin-dependent kinase-5 (CDK5)
small interfering RNA (siRNA)
Glycogen synthase kinase-3 beta (GSK3β)
Phosphotau (ptau)
Human tau (hTau)
NLR family pyrin domain-containing 3 (NLRP3)
Nuclear factor kappa B (NF-κB)
Mild cognitive impairment (MCI)
Neurofibrillary tau tangles (NFT)
Frontotemporal dementia with parkinsonism-17 (FTD-17)
Progressive supranuclear palsy (PSP)
Parkinson’s disease (PD)
European Medicines Agency (EMA)
Toll-like receptors (TLRs)
Acetylcholine esterase (AChE)
Reactive oxygen species (ROS)
Intracerebroventricular (ICV)
Huntington’s disease (HD)
Cytosine–adenine–guanine (CAG)
Mutant huntingtin (mHtt)
Huntingtin protein (Htt)
Medium spiny neurons (MSNs)
Associated speck-like protein (ASC)
Voltage-gated anion channels (VRAC)
Novel object recognition (NOR)
Voltage-gated sodium channels (VGSC)
Mithramycin (MTM)
Anti-seizure drugs (ASD)
GABA transporter 1 (GAT)
Author Contributions: Conceptualization: N.Z. and J.H.; writing—original draft preparation: J.H.; writing—review, and editing: J.H. and N.Z. All authors have read and agreed to the published version of the manuscript. * Figures 2, 3, and the abstract graphic were created using BioRender.com accessed August 2020.
Funding: This research received no external funding.
Institutional Review Board Statement: Not applicable
Informed Consent Statement: Not applicable
Data Availability Statement: Data sharing not applicable.
Conflicts of Interest: The authors declare no conflict of interest.
This entry is adapted from the peer-reviewed paper 10.3390/cells10030702
References
- Joo, Y.; Kim, H.S.; Woo, R.S.; Cheol, H.P.; Shin, K.Y.; Lee, J.P.; Chang, K.A.; Kim, S.; Suh, Y.H. Mefenamic Acid Shows Neuroprotective Effects and Improves Cognitive Impairment in in Vitro and in Vivo Alzheimer’s Disease Models. Mol. Pharmacol. 2006, 69, 76–84.
- Daniels, M.J.D.; Rivers-Auty, J.; Schilling, T.; Spencer, N.G.; Watremez, W.; Fasolino, V.; Booth, S.J.; White, C.S.; Baldwin, A.G.; Freeman, S.; et al. Fenamate NSAIDs Inhibit the NLRP3 Inflammasome and Protect against Alzheimer’s Disease in Rodent Models. Nat. Commun. 2016, 7.
- DuBois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; Putte, L.B.A.; Lipsky, P.E. Cyclooxygenase in Biology and Disease. FASEB J. 1998, 12, 1063–1073.
- Fitzpatrick, F.A. Cyclooxygenase Enzymes: Regulation and Function. Curr. Pharm. Des. 2004, 10, 577–588.
- Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M.B.; Hawkey, C.J.; Hochberg, M.C.; et al. Comparison of Upper Gastrointestinal Toxicity of Rofecoxib and Naproxen in Patients with Rheumatoid Arthritis. N. Engl. J. Med. 2000, 343, 1520–1528.
- Patrono, C. Cardiovascular Effects of Cyclooxygenase-2 Inhibitors: A Mechanistic and Clinical Perspective. Br. J. Pharmacol. 2016, 82, 957–964.
- Hawkey, C.J. COX-1 and COX-2 Inhibitors. Best Pract. Res. Clin. Gastroenterol. 2001, 15, 801–820.
- Drini, M. Peptic Ulcer Disease and Non-Steroidal Anti-Inflammatory Drugs. Aust. Prescr. 2017, 40, 91–93.
- Krumholz, H.; Ross, J.S.; Presler, A.H.; Egilman, D.S. What Have We Learnt from Vioxx? BMJ 2007, 334, 120–123.
- Graham, G.G. Fenamates. In Compendium of Inflammatory Diseases; Parnham, M.J., Ed.; Springer, Birkhauser: Basel, Switzerland, 2016; pp. 1–6.
- Lees, P.; Giraudel, J.; Landoni, M.F.; Toutain, P.L. PK-PD Integration and PK-PD Modelling of Nonsteroidal Anti-Inflammatory Drugs: Principles and Applications in Veterinary Pharmacology. J. Veter Pharmacol. Ther. 2004, 27, 491–502.
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) and Organ Damage: A Current Perspective. Biochem. Pharmacol. 2020, 180, 114147.
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A Comprehensive Resource for in Silico Drug Discovery and Exploration. Nucleic Acids Res. 2006, 34, D668.
- Subaiea, G.M.; Alansi, B.H.; Serra, D.A.; Alwan, M.; Zawia, N.H. The ability of tolfenamic acid to penetrate the brain: A model for testing the brain disposition of candidate Alzheimer’s drugs using multiple platforms. Curr. Alzheimer Res. 2011, 8, 860–867.
- U.S. Food and Drug Administration. PONSTEL® (Mefenamic Acid Capsules, USP); U.S. Food and Drug Administration: Washington, DC, USA, 2008.
- Cimolai, N. The Potential and Promise of Mefenamic Acid. Expert Rev. Clin. Pharmacol. 2013, 6, 289–305.
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 Update: Improved Access to Chemical Data. Nucleic Acids Res. 2019, 47, D1102–D1109.
- Sostres, C.; Gargallo, C.J.; Arroyo, M.T.; Lanas, A. Adverse Effects of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs, Aspirin and Coxibs) on Upper Gastrointestinal Tract. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 121–132.
- Grosser, T.; Fries, S.; FitzGerald, G.A. Biological Basis for the Cardiovascular Consequences of COX-2 Inhibition: Therapeutic Challenges and Opportunities. J. Clin. Investig. 2005, 116, 4–15.
- Varga, Z.; Sabzwari, S.; Rafay, A.; Vargova, V. Cardiovascular Risk of Nonsteroidal Anti-Inflammatory Drugs: An Under-Recognized Public Health Issue. Cureus 2017, 9.
- Yu, Y.; Ricciotti, E.; Scalia, R.; Tang, S.Y.; Grant, G.; Yu, Z.; Landesberg, G.; Crichton, I.; Wu, W.; Puré, E.; et al. Vascular COX-2 Modulates Blood Pressure and Thrombosis in Mice. Sci. Transl. Med. 2012.
- Muraoka, S.; Miura, T. Inactivation of Cholinesterase Induced by Non-Steroidal Anti-Inflammatory Drugs with Horseradish Peroxidase: Implication for Alzheimer’s Disease. Life Sci. 2009, 84, 272–277.
- Etminan, M.; Gill, S.; Samii, A. Effect of Non-Steroidal Anti-Inflammatory Drugs on Risk of Alzheimer’s Disease: Systematic Review and Meta-Analysis of Observational Studies. BMJ 2003, 327, 128–131.
- Vlad, S.C.; Miller, D.R.; Kowall, N.W.; Felson, D.T. Protective Effects of NSAIDs on the Development of Alzheimer Disease. Neurology 2008, 70, 1672–1677.
- Meyer, P.F.; Tremblay-Mercier, J.; Leoutsakos, J.; Madjar, C.; Lafaille-Maignan, M.É.; Savard, M.; Rosa-Neto, P.; Poirier, J.; Etienne, P.; Breitner, J. A Randomized Trial of Naproxen to Slow Progress of Presymptomatic Alzheimer Disease. Neurology 2019, 92, E2070–E2080.
- Zhang, C.; Wang, Y.; Wang, D.; Zhang, J.; Zhang, F. NSAID Exposure and Risk of Alzheimer’s Disease: An Updated Meta-Analysis from Cohort Studies. Front. Aging Neurosci. 2018, 10, 83.
- 2020 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2020, 16, 391–460.
- Yassine, H.N.; Finch, C.E. APOE Alleles and Diet in Brain Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 150.
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339.
- Venkataraman, A.; Kalk, N.; Sewell, G.; Ritchie, C.W.; Lingford-Hughes, A. Alcohol and Alzheimer’s Disease-Does Alcohol Dependence Contribute to Beta-Amyloid Deposition, Neuroinflammation and Neurodegeneration in Alzheimer’s Disease? Alcohol Alcohol. 2017, 52, 151–158.
- Zhang, H.; Ma, Q.; Zhang, Y.; Xu, H. Proteolytic Processing of Alzheimer’s β-Amyloid Precursor Protein. J. Neurochem. 2012, 120 (Suppl. 1), 9–21.
- Tang, B.L. Amyloid Precursor Protein (APP) and GABAergic Neurotransmission. Cells 2019, 8, 550.
- Adwan, L.I.; Basha, R.; Abdelrahim, M.; Subaiea, G.M.; Zawia, N.H. Tolfenamic acid interrupts the de novo synthesis of the ?-amyloid precursor protein and lowers amyloid beta via a transcriptional pathway. Curr. Alzheimer Res. 2011, 8, 385–392.
- Abdelrahim, M.; Baker, C.H.; Abbruzzese, J.L.; Safe, S. Tolfenamic Acid and Pancreatic Cancer Growth, Angiogenesis, and Sp Protein Degradation. J. Natl. Cancer Inst. 2006, 98, 855–868.
- Subaiea, G.M.; Adwan, L.I.; Ahmed, A.H.; Stevens, K.E.; Zawia, N.H. Short-Term Treatment with Tolfenamic Acid Improves Cognitive Functions in Alzheimer’s Disease Mice. Neurobiol. Aging 2013, 34, 2421–2430.
- Adwan, L.; Subaiea, G.M.; Zawia, N.H. Tolfenamic Acid Downregulates BACE1 and Protects against Lead-Induced Upregulation of Alzheimer’s Disease Related Biomarkers. Neuropharmacology 2014, 79, 596–602.
- Leso, A.; Bihaqi, S.W.; Masoud, A.; Chang, J.K.; Lahouel, A.; Zawia, N. Loss in Efficacy Measures of Tolfenamic Acid in a Tau Knock-out Model: Relevance to Alzheimer’s Disease. Exp. Biol. Med. 2019, 244, 1062–1069.
- Adwan, L.; Subaiea, G.M.; Basha, R.; Zawia, N.H. Tolfenamic Acid Reduces Tau and CDK5 Levels: Implications for Dementia and Tauopathies. J. Neurochem. 2015, 133, 266–272.
- Brock, B.; Basha, R.; DiPalma, K.; Anderson, A.; Harry, G.J.; Rice, D.C.; Maloney, B.; Lahiri, D.K.; Zawia, N.H. Co-Localization and Distribution of Cerebral APP and SP1 and Its Relationship to Amyloidogenesis. J. Alzheimer’s Dis. 2008, 13, 71–80.
- Basha, M.R.; Wei, W.; Bakheet, S.A.; Benitez, N.; Siddiqi, H.K.; Ge, Y.W.; Lahiri, D.K.; Zawia, N.H. The Fetal Basis of Amyloidogenesis: Exposure to Lead and Latent Overexpression of Amyloid Precursor Protein and β-Amyloid in the Aging Brain. J. Neurosci. 2005, 25, 823–829.
- Zhang, H.; Wang, X.; Xu, P.; Ji, X.; Chi, T.; Liu, P.; Zou, L. Tolfenamic Acid Inhibits GSK-3β and PP2A Mediated Tau Hyperphosphorylation in Alzheimer’s Disease Models. J. Physiol. Sci. 2020, 70, 29.
- Chang, J.K.; Leso, A.; Subaiea, G.M.; Lahouel, A.; Masoud, A.; Mushtaq, F.; Deeb, R.; Eid, A.; Dash, M.; Bihaqi, S.W.; et al. Tolfenamic Acid: A Modifier of the Tau Protein and Its Role in Cognition and Tauopathy. Curr. Alzheimer Res. 2018, 15, 655–663.
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic Changes With Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381.
- Pontecorvo, M.J.; Keene, C.D.; Beach, T.G.; Montine, T.J.; Arora, A.K.; Devous, M.D.; Navitsky, M.; Kennedy, I.; Joshi, A.D.; Lu, M.; et al. Comparison of Regional Flortaucipir PET with Quantitative Tau Immunohistochemistry in Three Subjects with Alzheimer’s Disease Pathology: A Clinicopathological Study. EJNMMI Res. 2020, 10, 65.
- Caillet-Boudin, M.-L.; Buée, L.; Sergeant, N.; Lefebvre, B. Regulation of Human MAPT Gene Expression. Mol. Neurodegener. 2015, 10, 28.
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204.
- Dolan, P.J.; Johnson, G.V.W. The Role of Tau Kinases in Alzheimer’s Disease. Curr. Opin. Drug. Discov. Devel. 2010, 13, 595–603.
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 Inflammasome Activation Drives Tau Pathology. Nature 2019, 575, 669–673.
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328.
- Feng, X.; Fan, Y.; Chung, C.Y. Mefenamic Acid Can Attenuate Depressive Symptoms by Suppressing Microglia Activation Induced upon Chronic Stress. Brain Res. 2020, 1740, 146846.
- Shao, H.J.; Lou, Z.; Jeong, J.B.; Kim, K.J.; Lee, J.; Lee, S.H. Tolfenamic Acid Suppresses Inflammatory Stimuli-Mediated Activation of NF-ΚB Signaling. Biomol. Ther. 2015, 23, 39–44.
- Liu, X.; Li, Z.; Liu, H.; Zhu, Y.; Xia, D.; Wang, S.; Gu, R.; Wu, W.; Zhang, P.; Liu, Y.; et al. Low Concentration Flufenamic Acid Enhances Osteogenic Differentiation of Mesenchymal Stem Cells and Suppresses Bone Loss by Inhibition of the NF-ΚB Signaling Pathway. Stem Cell Res. Ther. 2019, 10, 1–14.
- Armagan, G.; Turunc, E.; Kanit, L.; Yalcin, A. Neuroprotection by Mefenamic Acid against D-Serine: Involvement of Oxidative Stress, Inflammation and Apoptosis. Free Radic. Res. 2012, 46, 726–739.
- Sukanya Jongsiriyanyong; Panita Limpawattana; Mild Cognitive Impairment in Clinical Practice: A Review Article. American Journal of Alzheimer's Disease & Other Dementiasr 2018, 33, 500-507, 10.1177/1533317518791401.
- Cai Gillis; Fariba Mirzaei; Michele Potashman; M. Arfan Ikram; Nancy Maserejian; The incidence of mild cognitive impairment: A systematic review and data synthesis. Alzheimer's & Dementia: Diagnosis, Assessment & Disease Monitoring 2019, 11, 248-256, 10.1016/j.dadm.2019.01.004.
- Eshkoor, S.A.; Mun, C.Y.; Ng, C.K.; Hamid, T.A. Mild cognitive impairment and its management in older people. Clin. Interv. Aging 2015, 10, 687–693, doi:10.2147/cia.s73922.
- Petersen, R.C. Mild Cognitive Impairment. Contin. Lifelong Learn. Neurol. 2016, 22, 404–418, doi:10.1212/con.0000000000000313.
- Petersen, R.C.; Lopez, O.; Armstrong, M.J.; Getchius, T.S.D.; Ganguli, M.; Gloss, D.; Gronseth, G.S.; Marson, D.; Pringsheim, T.; Day, G.S.; et al. Practice Guideline Update Summary: Mild Cognitive Impairment Report of Theguideline Development, Dissemination, and Implementation. Neurology 2018, 90, 126–135, doi:10.1212/WNL.0000000000004826.
- Hui-Yun Chang; Tzu-Kang Sang; Ann-Shyn Chiang; Untangling the Tauopathy for Alzheimer’s disease and parkinsonism. Journal of Biomedical Science 2018, 25, 1-11, 10.1186/s12929-018-0457-x.
- Kristen E. Pleil; Emily G. Lowery-Gionta; Nicole A. Crowley; Chia Li; Catherine A. Marcinkiewcz; Jamie H. Rose; Nora M. McCall; Antoniette M. Maldonado-Devincci; A. Leslie Morrow; Sara R. Jones; et al. Effects of chronic ethanol exposure on neuronal function in the prefrontal cortex and extended amygdala. Neuropharmacology 2015, 99, 735-749, 10.1016/j.neuropharm.2015.06.017.
- Fulton T. Crews; Dipak K. Sarkar; Liya Qin; Jian Zou; Nadka Boyadjieva; Ryan P. Vetreno; Neuroimmune Function and the Consequences of Alcohol Exposure. Alcohol Research: Current Reviews 2014, 37, 331-351, .
- Venugopalan Rajesh; Mohanan Mridhulmohan; Subramanian Jayaseelan; Palanivel Sivakumar; Vellaiyachamy Ganesan; Mefenamic Acid Attenuates Chronic Alcohol Induced Cognitive Impairment in Zebrafish: Possible Role of Cholinergic Pathway. Neurochemical Research 2018, 43, 1392-1404, 10.1007/s11064-018-2554-3.
- Li, Y.; Zhong, W.; Jiang, Z.; Tang, X. New progress in the approaches for blood–brain barrier protection in acute ischemic stroke. Brain Res. Bull. 2019, 144, 46–57, doi:10.1016/j.brainresbull.2018.11.006.
- Chen, Q.; Olney, J.W.; Lukasiewicz, P.D.; Almli, T.; Romano, C. Fenamates Protect Neurons against Ischemic and Excitotoxic Injury in Chick Embryo Retina. Neurosci. Lett. 1998, 242, 163–166, doi:10.1016/S0304-3940(98)00081-0.
- Mary G. George; Leah Fischer; Walter Koroshetz; Cheryl Bushnell; Michael Frankel; Jennifer Foltz; Phoebe G. Thorpe; CDC Grand Rounds: Public Health Strategies to Prevent and Treat Strokes. MMWR. Morbidity and Mortality Weekly Report 2017, 66, 479-481, 10.15585/mmwr.mm6618a5.
- Mary F. Lopez; David A. Sarracino; Amol Prakash; Michael Athanas; Bryan Krastins; Taha Rezai; Jennifer N. Sutton; Scott Peterman; Oksana Gvozdyak; Sherry Chou; et al. Discrimination of ischemic and hemorrhagic strokes using a multiplexed, mass spectrometry-based assay for serum apolipoproteins coupled to multi-marker ROC algorithm. PROTEOMICS – Clinical Applications 2012, 6, 190-200, 10.1002/prca.201100041.
- Hung Wen Lin; Reggie H. C. Lee; Michelle H. H. Lee; Celeste Y. C. Wu; Alexandre Couto E Silva; Harlee E. Possoit; Tsung-Han Hsieh; Alireza Minagar; Cerebral ischemia and neuroregeneration. Neural Regeneration Research 2017, 13, 373-385, 10.4103/1673-5374.228711.
- Parto S. Khansari; Robert F. Halliwell; Mechanisms Underlying Neuroprotection by the NSAID Mefenamic Acid in an Experimental Model of Stroke. Frontiers in Neuroscience 2019, 13, 64, 10.3389/fnins.2019.00064.
- Parto S. Khansari; Robert F. Halliwell; Evidence for neuroprotection by the fenamate NSAID, mefenamic acid. Neurochemistry International 2009, 55, 683-688, 10.1016/j.neuint.2009.06.014.
- Peng Liu; Yinjie Li; Wei Yang; Danyang Liu; Xuefei Ji; Tianyan Chi; Zhutao Guo; Lin Li; Libo Zou; Prevention of Huntington’s Disease-Like Behavioral Deficits in R6/1 Mouse by Tolfenamic Acid Is Associated with Decreases in Mutant Huntingtin and Oxidative Stress. Oxidative Medicine and Cellular Longevity 2019, 2019, 1-13, 10.1155/2019/4032428.
- P. McColgan; S. J. Tabrizi; Huntington's disease: a clinical review. European Journal of Neurology 2017, 25, 24-34, 10.1111/ene.13413.
- S. N. Illarioshkin; S. A. Klyushnikov; V. A. Vigont; Yu. A. Seliverstov; E. V. Kaznacheyeva; Molecular Pathogenesis in Huntington’s Disease. Biochemistry (Moscow) 2018, 83, 1030-1039, 10.1134/s0006297918090043.
- Maria Jimenez-Sanchez; Floriana Licitra; Benjamin R. Underwood; David C. Rubinsztein; Huntington’s Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harbor Perspectives in Medicine 2016, 7, a024240, 10.1101/cshperspect.a024240.
- Sarah B. Thomson; Blair R. Leavitt; Transcriptional Regulation of the Huntingtin Gene. Journal of Huntington's Disease 2018, 7, 289-296, 10.3233/jhd-180331.
- Beghi, E.; Giussani, G.; Sander, J.W. The Natural History and Prognosis of Epilepsy. Epileptic Disord. 2015, 17, 243–253, doi:10.1684/epd.2015.0751.
- Pearson-Smith, J.N.; Patel, M. Metabolic Dysfunction and Oxidative Stress in Epilepsy. Int. J. Mol. Sci. 2017, 18, 2365, doi:10.3390/ijms18112365.
- Christoph Helmstaedter; Juri-Alexander Witt; Epilepsy and cognition – A bidirectional relationship?. Seizure 2017, 49, 83-89, 10.1016/j.seizure.2017.02.017.
- Asher Peretz; Nurit Degani; Rachel Nachman; Yael Uziyel; Gilad Gibor; Doron Shabat; Bernard Attali; Meclofenamic Acid and Diclofenac, Novel Templates of KCNQ2/Q3 Potassium Channel Openers, Depress Cortical Neuron Activity and Exhibit Anticonvulsant Properties. Molecular Pharmacology 2004, 67, 1053-1066, 10.1124/mol.104.007112.
- Edward C. Cooper; Lily Y. Jan; M-Channels: Neurological Diseases, Neuromodulation, and Drug Development.. Archives of Neurology 2003, 60, 496-500, 10.1001/archneur.60.4.496.
- Jian-Fang Sun; Yi-Jia Xu; Xiao-Hua Kong; Yang Su; Zhan-You Wang; Fenamates inhibit human sodium channel Nav1.7 and Nav1.8. Neuroscience Letters 2019, 696, 67-73, 10.1016/j.neulet.2018.12.008.
- Hau-Jie Yau; Gytis Baranauskas; Marco Martina; Flufenamic acid decreases neuronal excitability through modulation of voltage-gated sodium channel gating. The Journal of Physiology 2010, 588, 3869-3882, 10.1113/jphysiol.2010.193037.
- Rebecca S. Y. Wong; Role of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) in Cancer Prevention and Cancer Promotion. Advances in Pharmacological Sciences 2019, 2019, 1-10, 10.1155/2019/3418975.
- Basha, R.; Ingersoll, S.B.; Sankpal, U.T.; Ahmad, S.; Baker, C.H.; Edwards, J.R.; Holloway, R.W.; Kaja, S.; Abdelrahim, M. Tolfenamic acid inhibits ovarian cancer cell growth and decreases the expression of c-Met and survivin through suppressing specificity protein transcription factors. Gynecol. Oncol. 2011, 122, 163–170, doi:10.1016/j.ygyno.2011.03.014.
- Sankpal, U.T.; Abdelrahim, M.; Connelly, S.F.; Lee, C.M.; Madero-Visbal, R.; Colon, J.; Smith, J.; Safe, S.; Maliakal, P.; Basha, R. Small Molecule Tolfenamic Acid Inhibits PC-3 Cell Proliferation and Invasion in Vitro, and Tumor Growth in Orthotopic Mouse Model for Prostate Cancer. Prostate 2012, 72, 1648–1658, doi:10.1002/pros.22518.
- Eslin, D.; Sankpal, U.T.; Lee, C.; Sutphin, R.M.; Maliakal, P.; Currier, E.; Sholler, G.; Khan, M.; Basha, R. Tolfenamic Acid Inhibits Neuroblastoma Cell Proliferation and Induces Apoptosis: A Novel Therapeutic Agent for Neuroblastoma. Mol. Carcinog. 2013, 52, 377–386, doi:10.1002/mc.21866.
- Li, D.; Hu, C.; Li, H. Survivin as a novel target protein for reducing the proliferation of cancer cells (Review). Biomed. Rep. 2018, 8, 399–406, doi:10.3892/br.2018.1077.
- Sama F. Sleiman; Brett C. Langley; Manuela Basso; Jill Berlin; L. Xia; Jimmy B. Payappilly; Madan K. Kharel; Hengchang Guo; J. Lawrence Marsh; Leslie Michels Thompson; et al. Mithramycin Is a Gene-Selective Sp1 Inhibitor That Identifies a Biological Intersection between Cancer and Neurodegeneration. The Journal of Neuroscience 2011, 31, 6858-6870, 10.1523/JNEUROSCI.0710-11.2011.
- Shelake, S.; Sankpal, U.T.; Paul Bowman, W.; Wise, M.; Ray, A.; Basha, R. Targeting Specificity Protein 1 Transcription Factor and Survivin Using Tolfenamic Acid for Inhibiting Ewing Sarcoma Cell Growth. Investig. New Drugs 2017, 35, 158–165, doi:10.1007/s10637-016-0417-9.
- Ferrante, R.J.; Ryu, H.; Kubilus, J.K.; Mello, S.D. ’; Sugars, K.L.; Lee, J.; Lu, P.; Smith, K.; Browne, S.; Flint Beal, M.; et al. Neurobiology of Disease Chemotherapy for the Brain: The Antitumor Antibiotic Mithramycin Prolongs Survival in a Mouse Model of Huntington’s Disease. J. Neurosci. 2004, doi:10.1523/JNEUROSCI.2599-04.2004.
- Chao Wei; Wei Zhang; Qiong Zhou; Chao Zhao; Ying Du; Qi Yan; Zhuyi Li; Jianting Miao; Mithramycin A Alleviates Cognitive Deficits and Reduces Neuropathology in a Transgenic Mouse Model of Alzheimer’s Disease. Neurochemical Research 2016, 41, 1924-1938, 10.1007/s11064-016-1903-3.
- Venkata Subba Rao Atluri; Sneham Tiwari; Melisa Rodriguez; Ajeet Kaushik; Adriana Yndart; Nagesh Kolishetti; Mohan Yatham; Madhavan Nair; Inhibition of Amyloid-Beta Production, Associated Neuroinflammation, and Histone Deacetylase 2-Mediated Epigenetic Modifications Prevent Neuropathology in Alzheimer’s Disease in vitro Model. Frontiers in Aging Neuroscience 2020, 11, 342, 10.3389/fnagi.2019.00342.
- Nathaniel S. Woodling; Damien Colas; Qian Wang; Paras Minhas; Maharshi Panchal; Xibin Liang; Siddhita D. Mhatre; Holden Brown; Novie Ko; Irene Zagol-Ikapitte; et al. Cyclooxygenase inhibition targets neurons to prevent early behavioural decline in Alzheimer's disease model mice.. Brain 2016, 139, 2063-81, 10.1093/brain/aww117.
- Arti Tyagi; Mohammad A. Kamal; Nitesh Kumar Poddar; Integrated Pathways of COX-2 and mTOR: Roles in Cell Sensing and Alzheimer’s Disease. Frontiers in Neuroscience 2020, 14, 693, 10.3389/fnins.2020.00693.
- Li-Kai Huang; Shu-Ping Chao; Chaur-Jong Hu; Clinical trials of new drugs for Alzheimer disease. Journal of Biomedical Science 2020, 27, 1-13, 10.1186/s12929-019-0609-7.
- Li, Z.Y.; Yin, Y.F.; Guo, Y.; Li, H.; Xu, M.Q.; Liu, M.; Wang, J.R.; Feng, Z.H.; Duan, X.C.; Zhang, S.; et al. Enhancing Anti-Tumor Activity of Sorafenib Mesoporous Silica Nanomatrix in Metastatic Breast Tumor and Hepatocellular Carcinoma via the Co-Administration with Flufenamic Acid. Int. J. Nanomed. 2020, 15, 1809–1821, doi:10.2147/IJN.S240436.
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538, doi:10.3389/fimmu.2019.02538.
- Ya-Shuo Feng; Zi-Xuan Tan; Lin-Yu Wu; Fang Dong; Feng Zhang; The involvement of NLRP3 inflammasome in the treatment of Alzheimer’s disease. Ageing Research Reviews 2020, 64, 101192, 10.1016/j.arr.2020.101192.
- Wolfgang Löscher; Heidrun Potschka; Sanjay M. Sisodiya; Annamaria Vezzani; Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharmacological Reviews 2020, 72, 606-638, 10.1124/pr.120.019539.
- Graeme J. Sills; Michael A. Rogawski; Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 2020, 168, 107966, 10.1016/j.neuropharm.2020.107966.
- Mark Manford; Recent advances in epilepsy. Journal of Neurology 2017, 264, 1811-1824, 10.1007/s00415-017-8394-2.
- José Castillo; María Isabel Loza; David Mirelman; José Brea; Miguel Blanco; Tomás Sobrino; Francisco Campos; A novel mechanism of neuroprotection: Blood glutamate grabber. British Journal of Pharmacology 2015, 36, 292-301, 10.1177/0271678x15606721.
- José Castillo; Beyond Glutamate Antagonists for Treatment of Ischemic Stroke: Blood Glutamate Grabbing. Journal of Neurology & Neuromedicine 2016, 1, 31-34, 10.29245/2572.942X/2016/3.1030.
- Maen Abdelrahim; Stephen Safe; Cyclooxygenase-2 Inhibitors Decrease Vascular Endothelial Growth Factor Expression in Colon Cancer Cells by Enhanced Degradation of Sp1 and Sp4 Proteins. Molecular Pharmacology 2005, 68, 317-329, 10.1124/mol.105.011825.