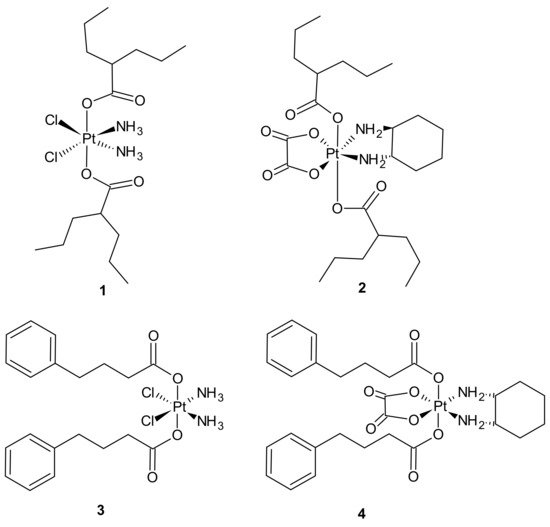
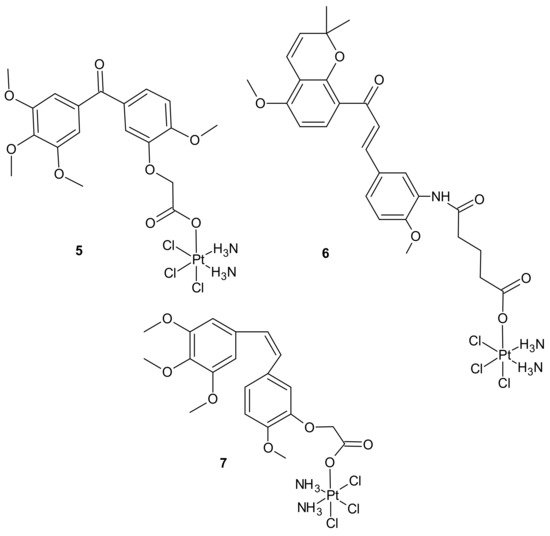
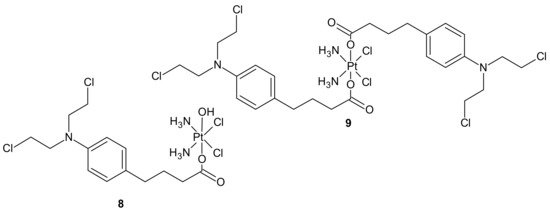
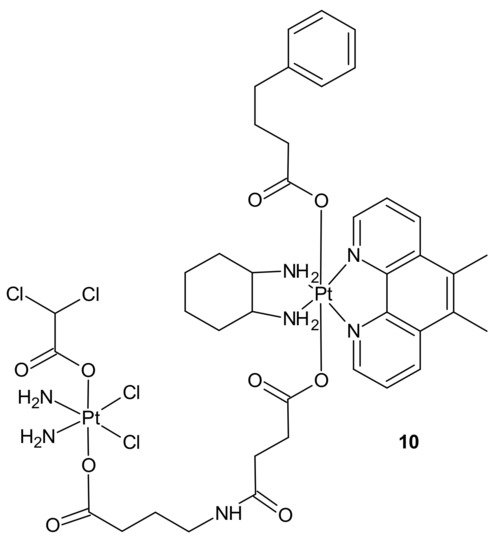
3.2.1. Drug Transporters, Drug Efflux, Drug Uptake
Multidrug resistance (MDR) is referred to as the ability of cancer cells to develop cross-resistance or insensitivity to many anticancer drugs, regardless of their chemical structure, mechanism of action, or molecular target [
75]. Drugs affected by multidrug resistance, e.g., vinca alkaloids, anthracyclines, or paclitaxel, are commonly used in a broad spectrum of chemotherapy regimens [
5,
76,
77,
78,
79]. Although cancer cells develop various mechanisms of resistance to cytotoxic drugs, the key trigger is overexpression of drug transporters from the ABC family in the cell membrane [
75]. ATP-binding cassette (ABC) transporters that utilize energy derived from ATP to mediate drug transport are a family of proteins involved in the uptake and efflux of xenobiotics. They play significant roles in the protection of normal cells and tissues by regulating biological membrane permeability [
79]. The three major ABC drug transporters—P-glycoprotein (P-gp or ABCB1), Multidrug Resistance Protein 1 (MRP1 or ABCC1), and ABCG2 (also known as MXR-mitoxantrone-resistance gene, BCRP-Breast Cancer Resistant Protein, or Placenta-specific ABC Transporter)—are most frequently associated with unfavorable clinical outcomes through the development of transporter-mediated MDR. They are capable of extruding a wide range of anticancer drugs, thus reducing their intracellular concentration and, subsequently, facilitating the development of resistance [
76].
P-glycoprotein (P-gp), a product of the ABCB1 (or MDR1) gene, is profoundly characterized and understood multidrug efflux transporter [
80,
81]. Since the overexpression of P-gp is one of the major problems hampering successful cancer chemotherapy, many agents have been designed and investigated for their ability to inhibit/modulate P-gp mediated MDR alone, or in combination with cytotoxic agents [
82,
83]. Nonetheless, the clinical outcomes are still disappointing due to their low potency and specificity. The need to use high doses results in serious side effects and toxicity to normal tissues, owing to the wide distribution of P-gp and its important physiological and pharmacological roles in the human body. The discovery of new and effective P-gp modulators without the abovementioned disadvantages is still a challenging task [
84]. Among various means of transient modulation of MDR, such as direct inhibition of ABC transporters, gene silencing, transcriptional regulation, and specific drug-delivery systems [
85], hybrid drugs, due to their multiple targeting nature, were also investigated as a promising way of overcoming P-gp-mediated MDR [
82].
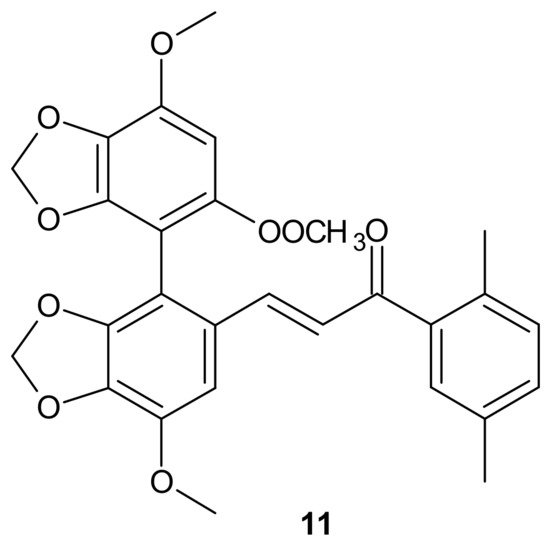
A series of bifendate–chalcone hybrids were synthesized and evaluated to enhance the P-gp inhibitory effect of bifendate. The most promising compound,
11 (), exerted little intrinsic cytotoxicity, whereas its chemosensitizing effect was prolonged and more potent than bifendate and verapamil (VRP). In addition,
11 () did not stimulate P-gp ATPase activity, which suggested that it is not a P-gp substrate [
86].
Figure 5. Structure of the most active bifendate-chalcone hybrid against P-gp function.
Another concept for overcoming P-gp-associated MDR was the design of hybrid drugs bearing an
N,
N-bis(alkanol)amine diester scaffold, combined with a coumarin or benzene sulfonamide moieties, with a synergistic inhibitory activity mechanism on P-gp and carbonic anhydrase [
87].
Carbonic anhydrase XII (CA XII) is a membrane enzyme that maintains suitable intra- and extracellular pH in tumor cells. It is highly expressed in some chemoresistant P-gp-positive cancer cells. Since optimal pH for efficient P-gp efflux activity in cancer cells is slightly alkaline, CA XII activity is critical for P-gp efflux. As a consequence, the activity of P-gp can be modulated by CA XII inhibition, producing a significant decrease in P-gp ATPase activity [
88,
89].
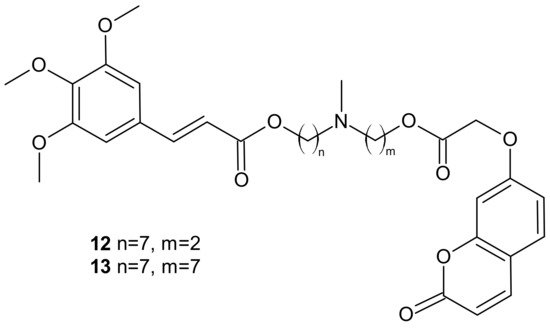
Among the tested compounds, derivatives bearing coumarin scaffolds
12 and
13 (), incorporating an (
E)-3-(3,4,5-trimethoxyphenyl)vinyl moiety linked to a 7-methylenes spacer, showed the best synergistic MDR inhibitory profile, being more potent on LoVo/DOX colon cancer cells that overexpressed both P-gp and CA XII than on K562/DOX leukemia cells overexpressing only P-gp. It made them potentially selective chemosensitizers able to overcome P-gp-mediated MDR with a synergistic antitumor mechanism [
87].
Figure 6. Selected dual P-gp and CA XII inhibitors.
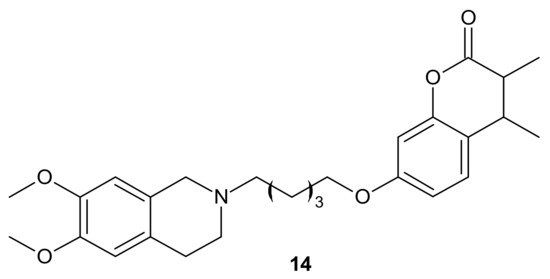
Rullo et al., obtained conjugates consisting of 1,2,3,4-tetrahydroisoquinoline (THIQ) moiety linked to coumarin scaffold through differently shaped and sized spacers. Compounds were evaluated in Madin-Darby Canine Kidney (MDCK) cells overexpressing P-gp and MRP1. Several THIQ-coumarin conjugates were identified as nanomolar P-gp inhibitors, and the most potent compound,
14, depicted in with a pentamethylene linker, showed nanomolar inhibition potency. In addition, it was elucidated that linker length and flexibility affected P-gp inhibition potency, whereas bulky groups (Br, Phenyl) at coumarin C3 improved P-gp/MRP1 selectivity. Some novel compounds could reverse the resistance of MDCK-MDR1 cells to doxorubicin [
90].
Figure 7. The most potent THIQ-coumarin conjugate.
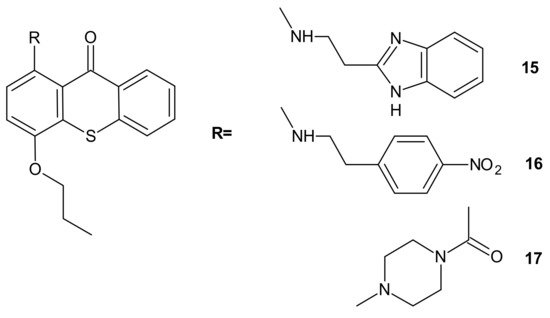
Hybrid compounds merging a thioxanthonic scaffold and an amine group, described as important pharmacophoric features for P-gp inhibition, were designed using homology modeling and docking by Palmeira et al. [
91]. 1-[2-(1
H-benzimidazol-2-yl)ethanamine]-4-propoxy-9
H-thioxanthen-9-one
15 () was identified as a potent P-gp inhibitor. It caused an accumulation rate of rhodamine-123 similar to verapamil (a known P-gp inhibitor) in the K562Dox resistant cell line. At a concentration of 10 µM, compound
15 caused a significant decrease in the GI
50 (growth inhibitory activity) value of doxorubicin in the K562Dox cell line, being 2-fold more potent than verapamil. Compounds
16 and
17 () at 10 µM sensitized resistant K562Dox cells (despite the lower rhodamine-123 accumulation in comparison to verapamil), probably due to their dual activity as P-gp and cell growth inhibitors [
91].
Figure 8. Structures of selected 1-aminated thioxantones.
In some cases, direct interaction of a hybrid drug with the nuclear target can prevent the excretion of the drug from the cell. Hybrids of combrestatin A (CA-4) and pironetin are a fine example [
92]. CA-4 is a plant-derived product which exerts its action by binding to ß-tubulin, at colchicine-binding site [
93,
94,
95], while pironetin (5,6-dihydro-α-pyrone of fungal origin) can bind to α-tubulin [
96,
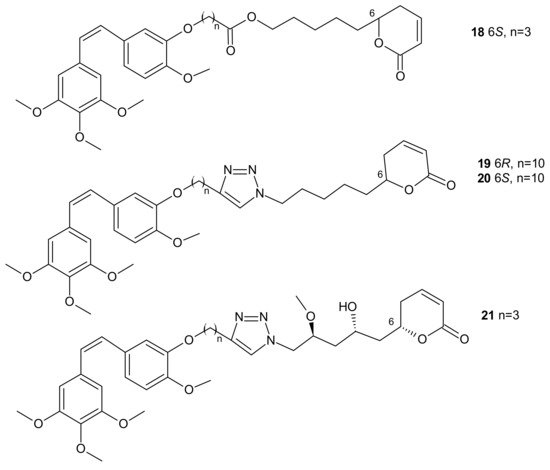
97]. Conjugates of CA-4 and pironetin, tethered by an ester-type linker of variable length or a 1,2,3-triazole spacer, were designed to display a dual affinity to both tubulin units. They could bind covalently to tubulin, which prevented the efflux pump from expelling them out of cells. Evaluation against two human ovarian carcinoma cell lines, A2780 (sensitive to chemotherapy) and A2780AD (resistant to chemotherapy), revealed the highest cytotoxicity for three compounds:
18,
19, and
20 (). The resistance factor (RF—the ratio of the IC
50 for A2780AD to IC
50 for A2780) for most of the compounds under study was close to unity, indicating that hybrid molecules of CA-4 and pironetin were also cytotoxic to multidrug resistant A2780AD cells. The effect of hybrid molecules of CA-4 and pironetin on the cell cycle was also evaluated in a non-small-cell lung adenocarcinoma cell line (A-549). Among tested compounds, two conjugates, namely
18 containing ester-type linker and
21 containing triazole linker (), exerted the highest antiproliferative activity against A-549 cell line. The same molecules displayed the strongest effect on depolymerization of the microtubule network [
92].
Figure 9. Representative combrestatin A (CA-4) and pironetin hybrid molecules.
Resistance related to drug efflux is common but, sometimes, a lack of therapeutic activity may be associated with impaired carrier-mediated transport of specific drugs into the cancer cell [
98].
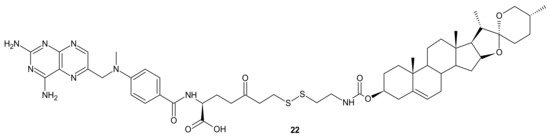
A deficiency in cancer of a reduced folate carrier mediated transport is the primary and frequent mechanism of resistance to methotrexate (MTX), which limits its clinical use as an anticancer drug [
99]. To overcome folate transporter-related resistance, a series of methotrexate-diosgenin conjugates with various polyamine linkers was designed [
100]. Diosgenin is a natural, strongly hydrophobic steroidal saponin, with a cholesterol-like structure exhibiting high biocompatibility and anticancer activity [
101]. MTX-diosgenin conjugates exhibited a stronger antiproliferative activity against transport-resistant breast cancer cell line MDA-MB-321 than MTX. They had the capability of entering MTX-resistant cells and retained the ability to inhibit dihydrofolate reductase (DHFR), despite diosgenin substitution. Conjugate
22 () possessing disulfide bond was the most active among obtained compounds [
100].
Figure 10. The most potent MTX-diosgenin conjugate.
Designing drug delivery systems is often reported as an efficient approach to overcome the efflux phenomena of anticancer drugs [
102], but the use of polymeric drug carriers is sometimes associated with a number of obstacles, such as low drug loading, side effects connected with degradation, metabolism, and excretion, as well as poor quality control [
103,
104]. The carrier-free, drug self-delivery system for cancer therapy proposed by Huang P. et al., seems to be an innovative solution [
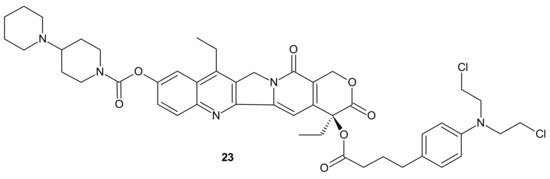
105]. An amphiphilic drug–drug conjugate (ADDC)
23 (), consisting of the hydrophilic anticancer drug irinotecan (Ir) tethered by a hydrolyzable ester linkage to the hydrophobic anticancer drug chlorambucil (Cb), exhibited the ability to assemble into nanoparticles, with longer blood retention half-life in comparison to free parent drugs. It facilitated the accumulation of drugs in tumor tissues and promoted subsequent cellular internalization, helping to avoid P-gp-mediated multidrug resistance. After ADDC
23 () hydrolysis, two released anticancer drugs exerted synergistic cytotoxicity to the tumor cells, exhibiting a higher apoptotic rate and anticancer activity than the individual free drugs. These advantages of Ir−Cb ADDC nanoparticles result in superior anticancer efficacy in vivo [
105].
Figure 11. Amphiphilic drug−drug conjugate (ADDC) Ir-Cb, self-assembling into ADDC nanoparticles.
3.2.2. Drug Inactivation (Enzymatic Detoxification)
The glutathione S-transferases (GSTs) represent a family of detoxification enzymes that catalyze the conjugation of glutathione (GSH) to many exogenous and endogenous compounds, including carcinogens, drugs, and oxidative stress products. Several mechanisms, such as transcriptional activation, stabilization of either mRNA or protein, and gene amplification, are reported to be involved in the overexpression of GSTs [
106]. MDR related to GSTs overexpression is a consequence of the GSH-anticancer drug conjugates’ formation and their active efflux via ABC transporters, or the inhibition of the mitogen-activated protein kinases pathway (MAPKs) [
107]. Many anticancer agents, such as adriamycin, 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU), busulfan, carmustine, chlorambucil, cisplatin, cyclophosphamide, melphalan, or thiotepa, are potent substrates of GSTs [
108,
109], which affects their anticancer activity and lead to adverse toxicity effects. [
107,
110]. Conjugation of platinum-containing cancer drugs, such as cisplatin and oxaliplatin, with GSH leads to their recognition as substrates for ABC transporters and enhances their efflux from the cancer cells [
111].
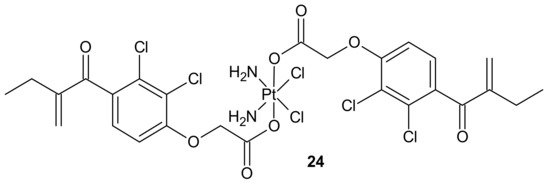
To overcome resistance to cisplatin associated with overexpression of π class glutathione S-transferase (GST π-1), ethacraplatin (EA-CPT)
24 (), a trans-Pt(IV) carboxylate complex containing ethacrynate ligands, was designed [
112]. Metallodrug
24 () strongly inhibited the GST activity in cell-free and cell systems (~10-fold of ethacrynic acid (EA) alone) and accelerated growth inhibition of cisplatin-resistant cancer cells, namely breast MCF-7 and T47D, lung A-549, and colon HT29 human carcinoma cells, in comparison to cisplatin [
113]. Unfortunately, in the case of malignant pleural mesothelioma (MPM) cells, compound
24 () could not inhibit cellular GST activity. Therefore, it was postulated that its activity may be related to the type of cancer [
114].
Figure 12. Structure of ethacraplatin (EA-CPT).
3.2.3. DNA Damage Repair
Many chemotherapeutic drugs, such as methylating, chloroethylating, and platinum-based agents commonly used for treating cancer, exhibit antitumor effects by causing direct DNA damage in tumor cells [
115]. Their effectiveness is often negatively influenced by the ability of cancer cells to repair this DNA damage. The role of DNA damage repair in drug resistance was excellently reviewed in [
116]. One of the commonly described mechanisms of resistance to alkylating agents is increased O
6-alkylguanine-DNA alkyltransferase (MGMT) protein activity, which repairs the cytotoxic O
6-methylguanine (O
6-BG) DNA adduct through direct transfer of the alkyl group to its cysteine residue [
117] and prevents its harmful effects, resulting in cell apoptosis [
118]. Since high levels of MGMT in tumor cells result in severe resistance to guanine O
6-alkylating agents, a series of methyltriazene hybrids bearing DNA methylating triazenes and O
6-BG was obtained. Hydrolysis of obtained compounds under physiological conditions released O
6-BG (a pseudosubstrate of MGMT) and DNA-damaging methyldiazonium derivatives, while
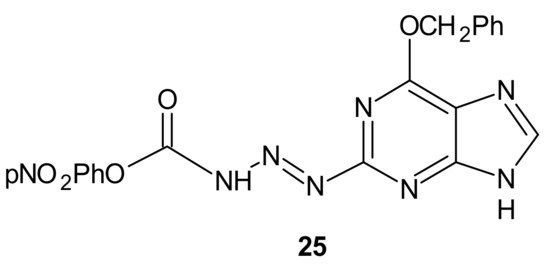
p-nitrophenyloxy derivative
25, depicted in , exhibited favorable penetration properties, and was the most active toward the NCI-60 panel of human tumor cell lines [
119].
Figure 13. The most potent methyltriazene hybrid bearing O6-bnzylguanine moiety.
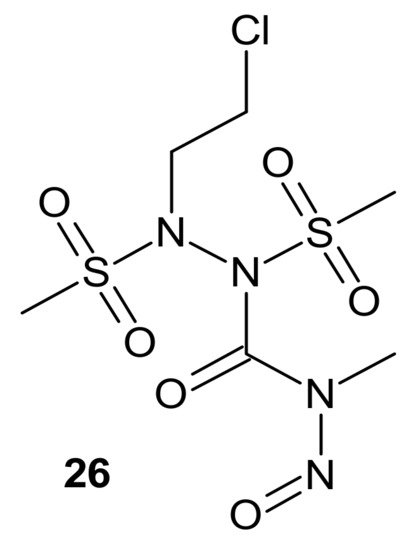
Zhu et al., obtained compounds with dual chloroethylating and methylating functions. Methylating moiety was introduced to deplete MGMT activity, which enhanced the sensitivity of cells to chloroethylating entities, inducing lethal interstrand cross-links. Compound
26 () exhibited the highest toxicity against human prostate cancer cell line DU-145, which expressed MGMT [
120].
Figure 14. A hybrid compound with dual chloroethylating and methylating functions.
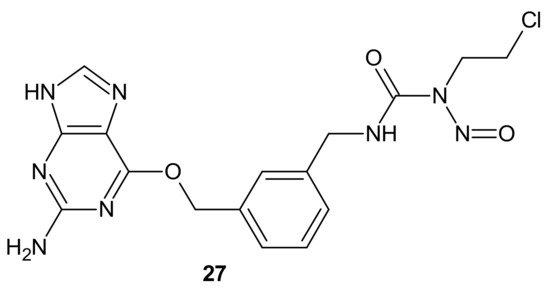
Combi-nitrosourea molecule
27 (), releasing a DNA cross-linking agent and an inhibitor of MGMT, was designed in response to cancer resistance to chloroethylnitrosoureas (CENUs) induced by MGMT, which repairs O
6-alkylated guanine and, subsequently, inhibits the formation of dG–dC cross-links. It hampers the application of CENUs in chemotherapy regimens. Molecule
27 () exhibited higher cytotoxicity against MGMT high-expressing glioma cells compared with nimustine, carmustine, and their respective combinations with O
6-BG. The results suggested that the superiority of
27 () is related to the simultaneous release of a chloroethyldiazonium ion, inducing DNA cross-link and O
6-BG analogs inhibiting the MGMT-mediated drug resistance in MGMT high-expressing cells [
121].
Figure 15. Representative combi-nitrosourea hybrid.
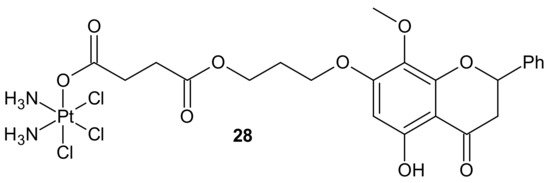
In addition to the previously mentioned hybrid Pt(IV) complexes (see
Section 3.1), several platinum hybrids, with bioactive ligands preventing multiple DNA damage response-mediated pathways, were designed [
66]. Cis-wogonin
28 () is an octahedral Pt (IV) conjugate containing wogonin (5,7-dihydroxy-8-methoxyflavone, isolated from
Scutellaria baicalensis, exhibiting multiple anticancer effects on gastric, lung, and glioma cancer cells [
122]) as an axial ligand. Cis-wogonin
28 (), designed to suppress DDR (DNA damage repair)-related proteins, could reverse existing cisplatin resistance. The significant antitumor activity of Cis-wogonin
28 () was related to its ability to suppress JWA (retinoic acid-induced cytoskeleton-like gene involved in base excision repair (BER) and DNA single-strand break repair (SSB) processes) and its multi-interaction with X-ray repair, cross complementing 1 protein (XRCC1) required for efficient repair of SSBs [
123]. Cis-wogonin
28 () seemed to be a promising cytotoxic agent reversing cisplatin resistance by targeting SSBs repair pathways and inducing apoptosis [
124].
Figure 16. Structure of Pt-wogonin conjugate.
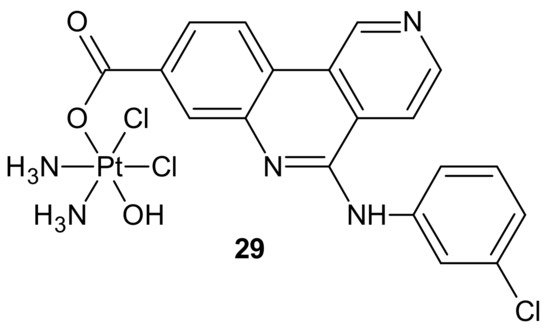
Another hybrid drug, Cx-platin
29, showed in , was designed to disrupt the DNA damage response mediated by Casein Kinase 2 (CK2). CK 2 is a constitutively active Ser/Thr protein kinase overexpressed in cancer cells. It is involved in regulating many prosurvival cellular processes, playing important roles in resistance to various chemotherapeutics [
125,
126,
127,
128,
129,
130] and the surveillance and repair of both single- and double-strand breaks [
131]. Cx-4945 is a selective ATP competitive inhibitor of CK 2, exhibiting activity against numerous malignancies in vitro and in vivo [
132,
133]. It was elucidated that the antiproliferative activity of Cx-4945 is related to modifications of signaling pathways such as PI3K/AKT, impairing DNA repair response, angiogenesis, splicing regulation, stress-induced cell death, or epigenetic modulation [
133,
134,
135,
136,
137,
138]. The inhibition of CK2 by Cx-4945 strongly enhanced the efficacy of cisplatin [
134]. Cx-platin
29 (), prepared by a fusion of cisplatin and Cx-4945 as an axial ligand, exhibited superior cytotoxicity in comparison to cisplatin against cancer cell lines, with distinct CK2-expressed levels, through suppressing CK2-mediated DNA damage repair and reversed cisplatin resistance. Due to high lipophilicity and complex stability,
29 () was highly accumulated in tumor cells. Moreover, subsequent profound platination of DNA caused severe DSB and SSB damage, inducing enhanced cell cycle arrest and apoptosis compared with cisplatin and the combination of cisplatin and CX-4945. Further in vivo tests showed that
29 () displayed high tumor inhibition rates and barely any toxicity effects in contrast to cisplatin [
139].
Figure 17. Structure of Cx-platin hybrid.
3.2.4. Target Modifications
Another important defense line of cancer cells is their ability to camouflage drug targets. Mutation, amplification, as well as over- or under-expression of target levels ultimately lead to drug resistance [
1]. Such a mechanism is observed in cases of many anticancer drugs, e.g., tyrosine kinases [
140] or antitubulin agents. The latter can be classified as microtubule stabilizers (taxanes and epothilones) that stimulate tubulin polymerization, or destabilizers (vinca alkaloids, colchicine) that inhibit tubulin polymerization [
141]. In rapidly proliferating cancer cells, both events lead to cell cycle arrest and apoptosis [
142]. Mutations in tubulin subunits, changes in the tubulin isotype composition of microtubules, and alterations in microtubule-regulatory proteins are, apart from P-gp engaged in drug efflux [
7,
143], main factors influencing antitubulin drug resistance [
141]. Growing scientific evidence shows that overexpression of βIII-tubulin is responsible for the taxane resistance appearing in ovarian cancer [
144,
145,
146,
147], as well as in other tumor types, such as lung, breast, and gastric cancers [
148,
149].
It was revealed that compounds binding at colchicine can circumvent β III-tubulin-mediated resistance, which indicates the significant importance of designing new agents targeting colchicine-binding sites in an attempt to overcome taxane resistance in refractory cancers [
150].
Zhang and co-workers, inspired by successful combinations of taxanes with receptor tyrosine kinase inhibitors (RTKi), reported on hybrid furo[2,3-
d]pyrimidines exhibiting potent dual antitubulin and antiangiogenic activities [
151].
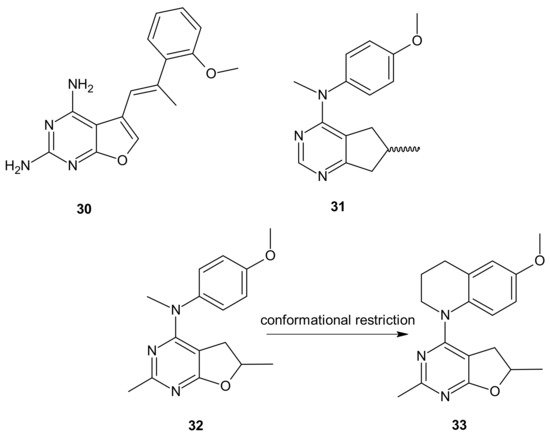
The choice of pharmacophores was inspired by the results of previous studies reporting that the compound
30 (), based on furo[2, 3 −
d]pyrimidine scaffold, had afforded both VEGFR-2 and PDGFR-β inhibitory activity [
152,
153], while 6-methyl cyclopenta-fused pyrimidines
31 () exhibited potent tubulin depolymerization activity, together with significant in vitro and in vivo antitumor activity [
154,
155,
156]. Among synthesized derivatives, compound
32 (), showed excellent dual RTK (VEGFR-2 and PDGFR-β) and microtubule inhibitory activity. In addition, it exhibited antiproliferative activity against the NCI-60 panel of cancer cells at low nanomolar levels, and was active in either paclitaxel-resistant tumor cell lines with βIII tubulin, or those that overexpressed P-gp. Moreover,
32 () showed potent growth inhibitory activity against tumor cells expressing VEGFR-2 and PDFGR-β, which is comparable to sunitinib, and caused cell cycle arrest in the G2/M phase, with subsequent apoptosis. The microtubule depolymerization through binding at the colchicine site was determined to be the primary mechanism of its antitubulin action. Compound
32 () also provided excellent antitumor activity superior to docetaxel and sunitinib in two murine models, reducing tumor size, vascularity, and metastases without obvious toxicity. An additional advantage of
32 () over clinical antimitotic agents, e.g., paclitaxel, is its simple synthetic procedure and the possibility of its conversion to the water-soluble salt form [
151].
Figure 18. Hybrid furo[2,3-d]pyrimidines exhibiting dual antitubulin and antiangiogenic activities.
The conformational restriction of
32 (), leading to the tetrahydroquinoline analog
33 (), enabled significant potency improvement against most of the NCI-60 panel cancer cells, including several chemoresistant cell lines. Compound
33 () showed potent RTK inhibition with nanomolar IC
50 value and exhibited microtubule depolymerizing activity comparable to or better than the lead compound
32 [
157].
Deregulation of tyrosine kinases (TK), being essential components of signaling cascades involved in diverse biological processes such as growth, differentiation, metabolism, and apoptosis, is connected with tumor proliferation, invasion, and metastasis, as well as tumor neovascularization. As a consequence, protein kinase inhibitors (TKIs) constitute important molecularly targeted drugs, broadly used for treating various malignancies [
158]. Their introduction into clinical practice was a real breakthrough and opened the way to different targeted therapies, significantly improving patient outcomes without the severe toxicity characteristic of commonly used chemotherapeutics [
159]. Unfortunately, the usefulness of TKIs is sometimes impaired by poor cancer response and acquired resistance. The predominant mechanism involved in resistance to TKI therapy, apart from gene amplification, overexpression, or alterations of signaling pathways, is the emergence of point mutations of the target kinase at the drug–kinase–interaction domain [
140,
158,
160].
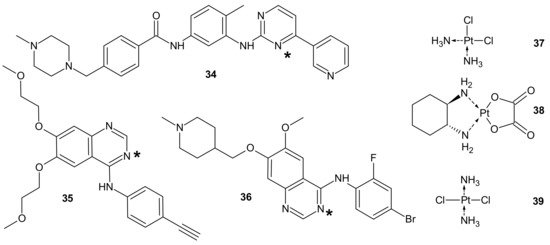
To circumvent TKI resistance, Pt-TKI hybrid compounds () were obtained by incorporating one of three TKIs (imatinib
34, erlotinib
35, and vandetanib
36) into the core structure of Pt-based anticancer drugs (cisplatin
37, oxaliplatin
38 and transplatin
39, ). Hybrids were formed by the reaction of Pt with the nitrogen atom on the quinazoline ring (erlotinib
35 and vandetanib
36) and pyrimidine ring (imatinib
34) of the TKIs. The additional hydrogen bonds facilitating the binding of the hybrids to altered sites made them less prone to resistance, causing secondary mutations. In addition, a dual anticancer mechanism of the Pt-TKI hybrids was observed, encompassing the inhibition of oncogenic kinases and monofunctional platination of DNA, which also probably contributed to the circumvention of drug resistance. The hybrids were not affected by the overexpression of the transporters in resistant cells and might be able to penetrate the blood–brain barrier to treat cancer metastasis in the brain. Apart from being effective in treating tumors bearing both sensitizing and resistance mutations, such as the third-generation mutant selective epidermal growth factor EGFR-TKI, the hybrids are also expected to exhibit a better side effect profile than the first generation TKIs [
161].
Figure 19. PT-TKI hybrids. Each TKI (34–36) was conjugated with each platinum derivative (37–39) through nitrogen atom labelled *.
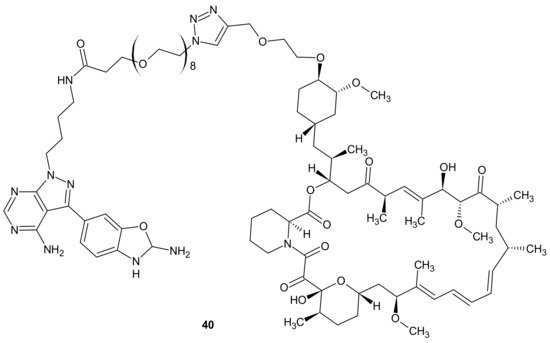
Mammalian Target Of Rapamycin Kinase (mTOR) mutations, such as mutations located in FRB-domain, as well as mutation located in kinase domain, are responsible for resistance to rapamycin analogs (rapalogs, e.g., temsirolimus, everolimus that are first in class mTOR kinase inhibitors approved for cancer therapy [
162]) and second generation mTOR ATP competitive inhibitors (TORKi), respectively [
163]. The exploitation of both ATP and FRB binding sites of mTOR generated the third class of mTOR inhibitors, represented by RapaLink-1
40 and depicted in . Compound
40 is a bivalent mTOR inhibitor consisting of rapamycin-FRB binding moiety, appropriately linked to the TORKi molecule. It prevents tumor growth in wild-type mTOR expressing cells and in cells with acquired resistance to the first and the second generation of mTOR inhibitors. A unique bivalent interaction of RapaLink-1
40 with mTOR is that binding at one site places the second half of the ligand in close proximity for binding to the second site, thus overcoming point mutations that diminish drug binding [
163]. In comparison with temsirolimus, which is the first-line therapy of renal cell carcinoma (RCC),
40 () showed significantly improved inhibition of proliferation, migration, invasion, and colony formation in the treatment of sunitinib-sensitive and sunitinib-resistant RCC cells in vitro and in vivo. It suppressed the mTOR signaling pathway, a part of the MAPK signaling pathway, the ErbB signaling pathway, and ABC transporters that are associated with resistance to many drugs [
164].
Figure 20. Structure of RapaLink-1.
3.2.5. Alterations in Signaling Pathways
Alterations in signaling pathways associated with survival (PI3K, AKT, mTOR, STAT), proliferation (MAPK, STAT, growth factors: FGFs, EGFR, IGFs), and cell death (BCL2 family and PTEN) are resistance mechanisms characteristic of targeted therapies [
6].
In case of multifactorial diseases such as cancer, the strong specific inhibition of a particular dysregulated pathway is insufficient to hamper the disease, because changes in signaling networks can reduce the effect of the drug through the activation of new aberrant cellular pathways. Moreover, targeted therapies often unintentionally provoke the preferential growth of resistant cells due to selective pressure on single elements of dysregulated pathways [
165].
The PI3K/AKT/mTOR is a common example of a hyper-activated signaling pathway in cancer cells. Phosphatidylinositol 3-Kinase (PI3K) and mTOR synergistically promote tumor progression and resistance to chemotherapy; therefore, inhibition of this pathway is an excellent treatment strategy and an efficient tool to avoid or circumvent cancer chemotherapy resistance [
166].
Rapalogs, aforementioned in
Section 3.2.4, are partial inhibitors of mTOR through allosteric binding to mTOR complex-1 (mTORC1), but not mTOR complex-2 (mTORC2) [
167,
168]. Since mTORC1 negatively regulates insulin/IGF-1 receptor signaling, mTORC1 blocking by rapalogs can stimulate PI3K/AKT signaling and antagonize their antitumor efficacy [
168,
169]. It was elucidated that treatment with rapamycin or rapalogs led to increased PI3K activity and AKT signaling, resulting in minimal inhibition of tumor cell growth [
170,
171]. Co-targeting mTOR and PI3K/AKT signaling prevented mTOR inhibition-initiated AKT activation and enhanced antitumor effects in vitro and in vivo [
172].
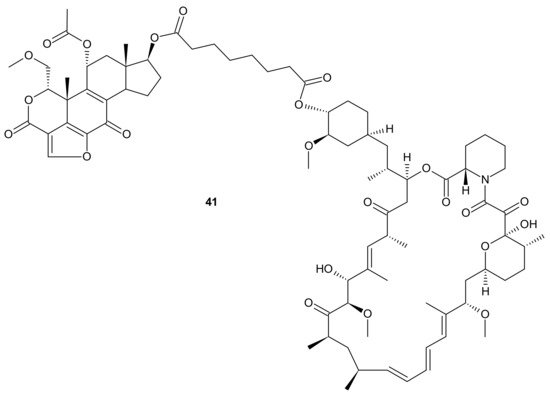
Kaloustian et al., conjugated 17-hydroxywortmannin (PI3K inhibitor) analog to rapamycin (mTOR inhibitor) through a diester linker which underwent hydrolysis in vivo and released two highly potent inhibitors of PI3K and mTOR. Conjugate
41 presented in , showed profound activity in the mouse glioma model (U87MG). In addition, at the dose of 15 mg/kg, compound
41 () inhibited the growth of human colon adenocarcinoma (HT29) tumors, whereas an equivalent mixture of rapamycin and 17-hydroxywortmaninn was poorly tolerated. In the A498 renal tumor model,
41 () exhibited superior efficacy over rapamycin or 17-hydroxywortmaninn when administered as a single agent or in combination with bevacizumab [
166].
Figure 21. Structure of dual PI3K and mTOR inhibitor with excellent activity in vivo.
Other examples of mTOR/PI3K dual inhibitors were extensively reviewed by Chen and Zhou [
173].
The activation of a signal transducer and activator of transcription 3 (STAT3) signaling pathway is present in a wide range of solid cancers and drug-resistant cancers in humans. It is associated with a worse prognosis [
174,
175,
176]. Therefore, inhibiting STAT3-mediated MDR1 gene expression is a possible treatment option for drug-resistant cancers [
176]. Zhang et al., proposed an interesting idea for overcoming cancer cell resistance in solid tumors by designing a new series of curcumin-BTP hybrids as STAT3 inhibitors with reactive oxygen species (ROS)-promoting activity [
177]. Curcumin and its analogs with improved anticancer potential and bioavailability inhibit numerous oncogenic processes, including those associated with the JAK2/STAT3 pathway or the induction of ROS production [
178,
179,
180,
181,
182,
183]. The benzo[b]thiophene 1,1-dioxide (BTP) moiety is a characteristic pharmacophore of many potent STAT3 inhibitors, markedly enhancing the ROS level and exhibiting antiproliferative activity in cancer cells [
184,
185]. Among obtained curcumin-BTP hybrids possessing drug-like properties and potent bioactivities in vitro and in vivo,
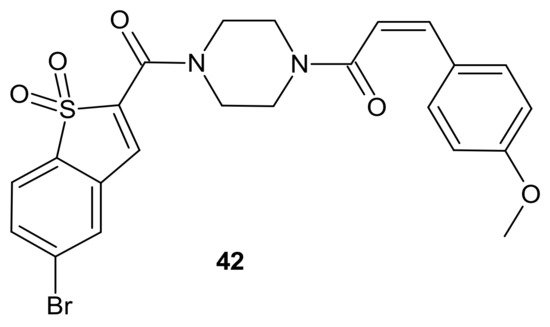
42 () exhibited the best antitumor activity and selectivity for MCF-7 and MCF-7/DOX cells, with significant STAT3 levels. Compound
42 () inhibited STAT3 activity, regulated the expression of STAT3 downstream genes Bcl-2, Bax and Cyclin D1, with little effect on p-Src or p-Erk, and also inhibited STAT3-mediated P-gp expression in MCF-7/DOX cells. In addition,
42 () was able to stimulate intracellular ROS production and accumulation. The aforementioned activity synergistically promoted cancer cell cycle arrest and apoptosis, which suggests that a combination of STAT3 inhibition with inducing high levels of ROS may be a valuable strategy to address resistance of cancer cells [
177].
Figure 22. Curcumin-BTP hybrid—a STAT3 inhibitor with reactive oxygen species (ROS)-promoting activity.
3.2.6. Epigenetic Alterations
The modulation of epigenetic processes is nowadays considered an innovative and interesting therapeutic strategy because dysfunctional gene regulation is responsible for many human diseases [
160] and emerging anticancer drug resistance [
3]. Epigenetic modifications in gene expression cannot be explained by changes in DNA base sequences. They are related to covalent modifications to the cytosine residues of DNA, histone covalent chemical modifications such as acetylation, methylation, etc., known as chromatin remodeling, and noncoding RNAs [
160]. Dysregulation of the dynamic status of histone acetylation plays an important role in the development of many malignancies [
186]. The acetylation of lysine catalyzed by histone acetyltransferases (HATs) decreases the affinity of histones for DNA and opens the chromatin structure to the more relaxed euchromatin form. It allows for the binding of various transcription factors to DNA and activates many downstream gene transcription processes. Deacetylation of lysine performed by HDAC enzyme leads to chromatin compaction and gene silencing [
187]. Therefore, HDAC inhibitors (HDACis) are epi-drugs that can reactivate signaling pathways silenced by deacetylation [
160], which, in turn, leads to an increase in the expression of tumor suppressors or decreases the expression of genes involved in tumorigenesis [
49]. HDACis exhibit multidirectional cellular effects, including the arrest of cell growth, cell cycle progression, and the induction of apoptosis. Moreover, through modification of both histone and non-histone substrates, they have the potential to disrupt multiple pathways of cancer resistance [
188]. It was elucidated that HDAC inhibitors, co-administrated with other anticancer agents, enhanced their anticancer effects, which was the inspiration for the design of HDACIs-based hybrid drugs [
50]. It must be underlined that the introduction of the pharmacophore for a second non-HDAC target would be impossible without a high degree of structural tolerance of the surface-binding cap, which enables the designing of multi-targeted molecules without compromising HDAC binding [
50]. The number of multi-target HDACi hybrids reported in the literature is overwhelming [
189,
190]. The examples described below show an undeniable and significant role of these hybrids in overcoming anticancer drug resistance.
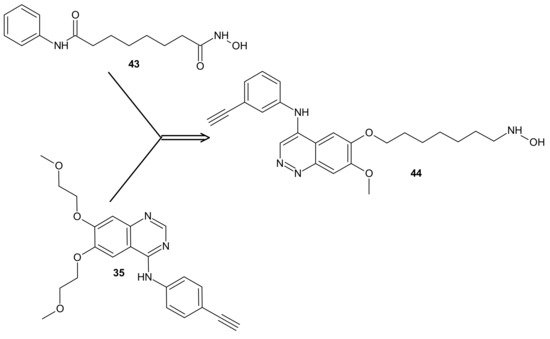
The design of RTKi/HDACi hybrids was proposed to overcome RTKi resistance, because combinations of HDACis and RTKis were shown to act synergistically [
191,
192,
193]. Such a hybrid consists of a key fragment from the kinase inhibitor, tethered by the appropriate linker to a zinc binding group from HDACi, as shown in [
194]. Many attempts were made to combine HDAC inhibitors and EGFR inhibitors in a single molecular entity [
55,
195,
196,
197].
Figure 23. CUDC-101 44 consists of a key fragment from the kinase inhibitor 35, tethered by an appropriate linker to a zinc binding group from HDACi-SAHA 43.
Cai et al., combined HDAC inhibitor vorinostat (SAHA)
43 () with EGFR inhibitor erlotinib
35 (), obtaining 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-
N-hydroxyheptanamide (CUDC-101)
44 () as a lead compound [
198]. CUDC-101
44 () is a multi-targeted entity that exhibited potent antiproliferative and proapoptotic activities against cancer cells in vitro and in drug-resistant tumor models in vivo. Cancer cells resistant to single-target EGFR inhibitors remain sensitive to
44 () [
199]. It was elucidated that
44 () integrated HDAC and EGFR/HER2 pathway inhibition, blocked and inhibited MET- and AXL-mediated signaling, and reduced cancer cell migration [
199,
200]. Phase I study reported good tolerance of
44 () and showed some preliminary evidence of its antitumor activity [
201].
Results of a multicenter phase I dose-escalation study in patients with locally advanced, intermediate-, or high-risk head and neck squamous cell cancer HNSCC also revealed good tolerance of
44 () in combination with cisplatin and radiation therapy. The maximum tolerated dose (MTD) of CUDC-101-based combination therapy was established at 275 mg/m
2/dose [
202].
Moreover, it was found that
44 () had potent antiproliferative and proapoptotic activities in anaplastic thyroid cancer (ATC) cell lines, with gene mutations in both the MAPK and the PI3K/AKT pathways [
203]. ATC belongs to the most lethal human malignancies [
204]. Treatment with
44 () reduced the expression of survivin, XIAP, N-cadherin, vimentin, β-catenin, and restored p21, as well as E-cadherin expression in ATC cells, which could also explain reduced tumor growth, metastasis, and prolonged survival observed in metastatic ATC mouse model in vivo [
203].
In addition,
44 () exerted significantly stronger antiproliferative effects than arsenic trioxide (ATO) in Acute Promyelocytic Leukemia (APL) and ATO-resistant APL cell lines, whereas it has a negligible cytotoxic effect on non-cancerous cell lines, including normal CD34+ cells and BMSCs from APL patients. Mechanistic studies revealed that
44 () directly induced hyperacetylation of histone 3, which led to the activation of caspase 3 and the degradation of the promyelocytic leukemia-retinoic acid receptor α (PML-RARα) fusion protein, which subsequently facilitated the apoptosis of APL and ATO-resistant APL cells. Moreover,
44 () significantly repressed leukemia development in vivo in the NB4 xenograft mouse model compared with ATO. These results suggested that
44 () could be a potential candidate drug for APL, particularly for ATO-resistant APL [
205,
206].
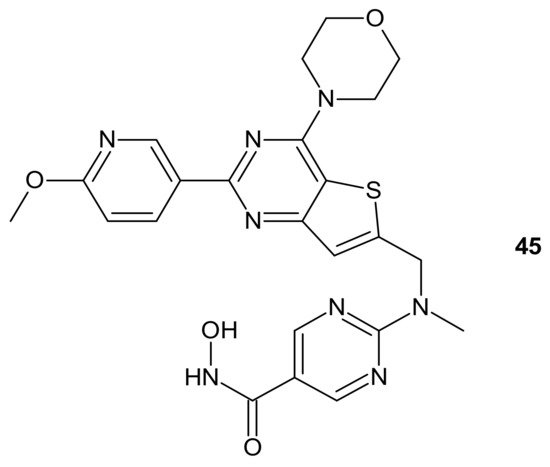
CUDC-907 (Fimepinostat)
45 () is an orally available hybrid compound consisting of hydroxamic acid representing HDAC inhibitory functionality, tethered to a morpholinopyrimidine moiety—the core structure of the PI3K inhibitors. It is a nanomolar inhibitor of HDACs (I, II, Iv classes), as well as four I class kinases PI3K [
207]. It () exhibited the potential to overcome drug resistance by simultaneous inhibition of PI3K activity and the disruption of cancer networks through epigenetic regulation of HDACs. It was elucidated that
45 () inhibited PI3K/AKT, JAK/STAT, and mitogen-activated protein kinase (MAPK) activation, as well as decreased C-MYC protein levels in solid tumor and hematological cell lines [
208,
209]. Moreover, the multidimensional modulation of dysregulated cancer pathways resulted in the significant antitumor activity of
45 () in many preclinical models of hematological malignancies and solid tumors [
209,
210,
211,
212,
213,
214], as well as clinical efficacy in relapsed/refractory lymphoma and multiple myeloma (MM) [
215,
216], MM, lymphoma, and advanced/relapsed solid tumors (newly diagnosed diffuse intrinsic pontine glioma (DIPG), recurrent medulloblastoma, or recurrent high-grade glioma (HGG)) in young adults and children [
217,
218].
Figure 24. Structure of CUDC-907 (Fimepinostat).
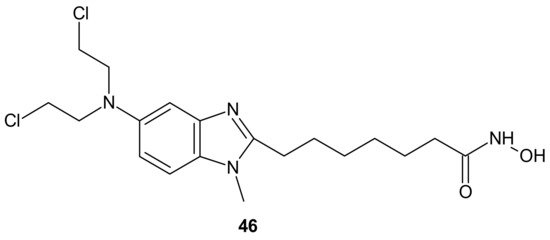
An alkylating histonedeacetylase inhibiting molecule, EDO-S101 (Tinostamustine)
46 (), obtained by the fusion of bendamustine with vorinostat, is the last example of a hybrid molecule described in our review. The rationale of its design was the synergy of HDAC-inhibitors and DNA-damaging agents relying on the HDACi-mediated chromatin relaxation that would make DNA more accessible to the damaging effect of bendamustine. EDO-S101
46 () exhibited simultaneous alkylating activity and HDAC inhibition in vitro and in vivo [
219]. Moreover, it showed significant activity against MM, leukemia, and B-cell lymphomas in preclinical models, with a toxicity profile similar to bendamustine [
220]. It () was the only drug to exhibit activity as a single agent in the multidrug resistant, transplant model of relapsed/refractory MM [
221]. This is in line with its outstanding synergistic cytotoxicity in combination with proteasome inhibitors against MM and a wide variety of B-cell neoplasms in vitro [
222]. EDO-S101
46 () is in early phase clinical development for a range of relapsed/refractory hematological malignancies, advanced solid tumors (small-cell lung cancer, soft tissue sarcoma, triplenegative breast cancer, ovarian cancer, endometrial cancer), and refractory, locally advanced or metastatic melanoma (with Nivolumab) [
218]. In addition, it exhibits potent radio-sensitizing activity in aggressive and temozolomide-resistant glioblastoma tumor models, supporting ongoing clinical evaluation of this compound in combination with radiotherapy for the treatment of post-surgery glioblastoma patients [
223]. It is well worth noting that the European Commission (EC) has adopted the recommendation of the European Medicines Agency (EMA) Committee for Orphan Medicinal Products to grant Orphan Drug Designation (ODD) to Tinostamustine
46 () for the treatment of T-cell prolymphocytic leukemia (T-PLL). The EC decision followed the FDA decision, which granted Tinostamustine ODD status for the treatment of T-PLL in March 2019 [
224].
Figure 25. Structure of EDO-S101 (Tinostamustine).