The cardiac conduction system is an extended network of excitable tissue tasked with generation and propagation of electrical impulses to signal coordinated contraction of the heart. The fidelity of this system depends on the proper spatio-temporal regulation of ion channels in myocytes throughout the conduction system. Importantly, inherited or acquired defects in a wide class of ion channels has been linked to dysfunction at various stages of the conduction system resulting in life-threatening cardiac arrhythmia. Members of the ankyrin and spectrin families have emerged as important nodes for normal expression and regulation of ion channels in myocytes throughout the conduction system. Human variants impacting ankyrin/spectrin function give rise to a broad constellation of cardiac arrhythmias. Furthermore, chronic neurohumoral and biomechanical stress promotes ankyrin/spectrin loss of function that likely contributes to conduction disturbances in the setting of acquired cardiac disease.
- pacemaking
- arrhythmia
- ankyrin
- spectrin
1. The Cardiac Conduction System in Health and Disease
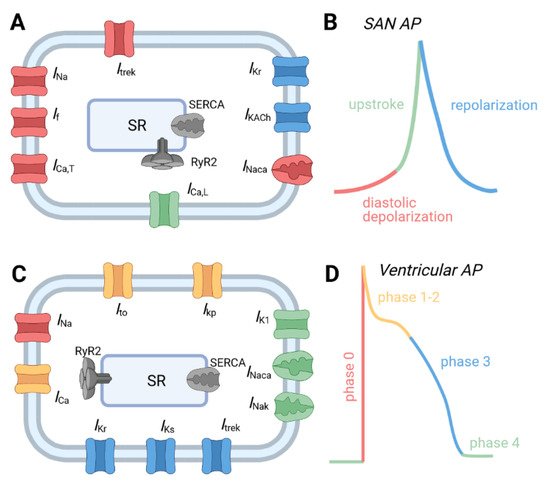
1.1. Normal Physiology of the Cardiac Conduction System
1.2. Cardiac Conduction System Dysfunction and Disease
2. Ankyrins in the Cardiac Conduction System
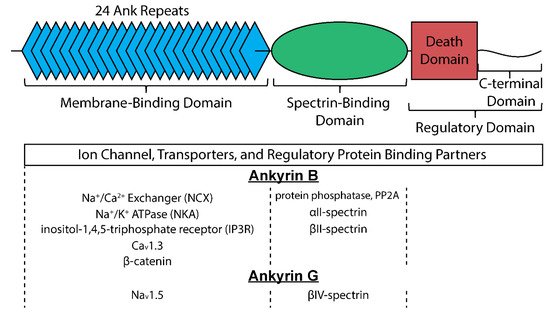
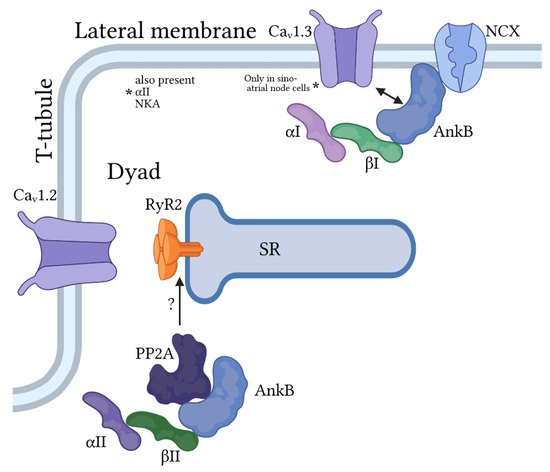
2.1. Ankyrin-B
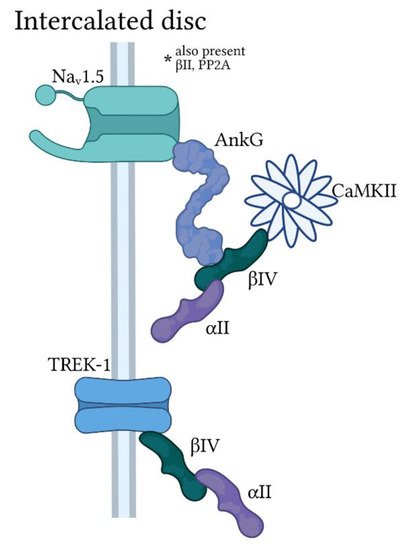
2.2. Ankyrin-G
3. Spectrins in the Cardiac Conduction System
3.1. αII-Spectrin
3.2. βII-Spectrin
3.3. βIV-Spectrin
This entry is adapted from the peer-reviewed paper 10.3390/jcdd8050048