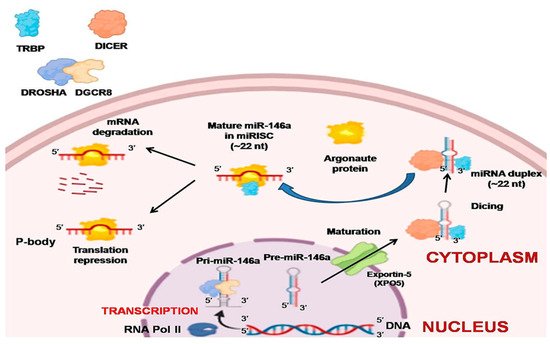
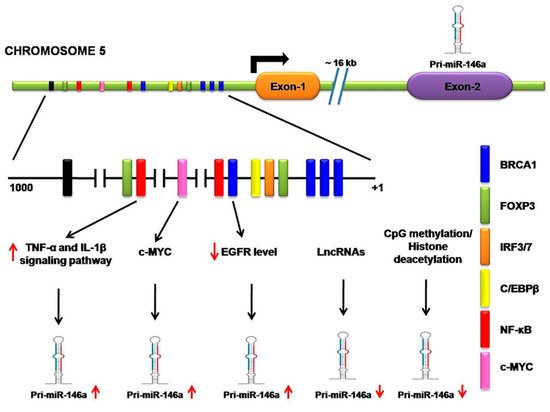
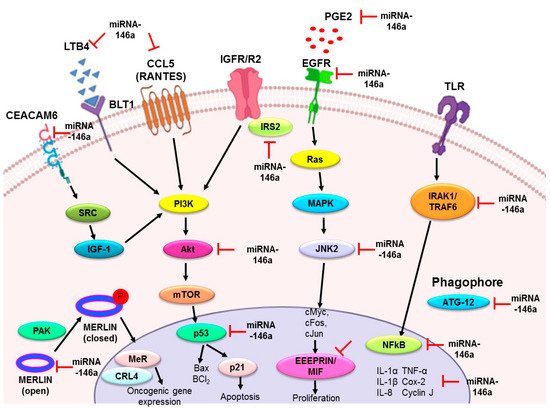
Most of the research associated with miR-146a indicates its tumor-suppressive role in malignancies. In NSCLC cell lines and human lung tissue samples, the expression of this miRNA is strongly downregulated and it also displays antiproliferative and antiapoptotic properties in lung cancer cell lines [
52,
53]. The gene for EGFR found to be commonly mutated in lung adenocarcinoma patients, especially in nonsmoking women with Asian ethnicity (50%) [
54,
55], is directly targeted by miR-146a [
19]. Qi et al. (2019) recently found that miR-146a-5p directly targets EGFR mRNA while studying the effect of cryptotanshinone derived from
Salvia miltiorrhiza on NSCLC. It was also observed that cryptotanshinone prevents cell cycle progression by upregulating miR-146a/b levels [
56]. The tumor collagenase stimulatory factor, also known as the extracellular matrix metalloproteinase inducer (EMMPRIN), is expressed on the outer surface of human tumor cells. This glycoprotein interacts with the fibroblasts and triggers many matrix metalloproteinase expressions within the fibroblasts [
57]. Huang WT et al. proved through various study methods like TargetScan, luciferase enzyme assay, cancer genome atlas, and immunohistochemistry that miR-146a directly targeted the tumor collagenase stimulatory factor (TCSF) to modulate the mechanisms of cell viability and apoptosis in NSCLC [
58]. Macrophage migration inhibitory factor (MIF) was considered an important cytokine for the regulation of innate immunity. Thorsten Hagemann et al. knocked down either EMMPRIN or MIF and found decreased invasiveness and matrix metalloproteinase activity in the supernatant of tumor cell culture [
59]. MIF was also found to be the promoter of the Warburg effect in NSCLC by activating NF-kB/HIF-1α inflammatory signaling [
60,
61]. The macrophage migratory inhibition factor (MIF) gene upregulated in NSCLC is the reverse target of miR-146a, as proven by luciferase assay mimic studies in A549 cells, and promotes apoptosis and discourages proliferation [
62]. Another similar study found cell cycle progression was also slowed down by miR-146a via specifically downregulating the expression of CCND1/2 (genes of cyclins D1/2), both at post-transcriptional and protein stages that arrested the G0/G1 phase of cell cycle progression [
63] and targeted cyclin J that promoted NSCLC chemosensitivity to cisplatin [
64]. It was found that miR-146a overexpression maintained epithelial phenotypes and discouraged epithelial to mesenchymal transition in lung cancer cell lines by suppressing insulin receptor substrate-2 (IRS2) transcription and translation [
65]. It has also been found that IRS2 enhances the Wnt/β-catenin pathway, thus increasing N-cadherin but decreasing E-cadherin [
66]. This was further confirmed by a study of a xenograft mouse model where miR-146a overexpressing cells decreased the tumor sizes. Downexpression of miR-146a was recently found to be caused by some epigenetic cellular events like promoter hypermethylation [
20] and post-transcriptional silencing by competitive endogenous RNA (ceRNA) in NSCLC. It was also recently found that SNHG16, a competitive endogenous RNA (ceRNA), was associated with poor prognosis and upregulation in NSCLC [
43]. However, the above observations were contradicted by Tan et al., who observed miR-146a overexpression both in vivo and in vitro experimental systems such as malignant lung tissue and NSCLC cell lines. He experimentally justified miR-146a as an oncomer since CHOP (DNA damage-inducible transcript 3) was targeted by miR-146a, whose downexpression has been linked to poor prognosis in lung cancer [
67]. The Merlin/NF2 tumor suppressor protein level was proven to be negatively regulated by miR-146a by directly interacting with its mRNA and triggering cell proliferation, invasion, and cell migration by miR-146a in vivo cell transfection studies in A549 lung epithelial cells [
68]. However, a large body of evidence proved the tumor-suppressive role of miR-146a in NSCLC.