1. Unfolded Protein Response (UPR)
As an ultimate perinuclear organelle, the endoplasmic reticulum (ER) is a membranous labyrinth network where cell-surface and secreted proteins can be synthesized and maintained with high fidelity through the assistance of molecular chaperones (e.g., glucose-regulated protein 78 kD or immunoglobulin heavy chain-binding protein, GRP78/BiP) and folding enzymes (e.g., protein disulfide isomerases, PDI) [
1,
2]. Only correctly folded proteins can be transported to the Golgi apparatus. Unfolded or misfolded proteins are retained in the ER and further inversely translocated from the ER lumen to the cytosol by a cellular procedure called the “Endoplasmic-Reticulum-Associated Protein Degradation (ERAD)”. ERAD designates a cellular pathway that targets unfolded or misfolded proteins in the ER for ubiquitination and subsequent degradation, usually by the 26S proteasome [
1,
2].
An imbalance between the load of misfolded protein generated in the ER and ERAD machinery triggers a series of cytoprotective signaling pathways called the Unfolded Protein Response (UPR) [
1,
2,
3]. Upon the onset of ER stress, GRP78 dissociates from its binding partners: Inositol-requiring enzyme 1α and β (IRE1α and β), activating transcription factor-6α (ATF-6α) and pancreatic ER kinase (PERK). Dissociation of GRP78 from these three complexes activates the protective UPR [
1,
2,
3]. The UPR plays four main functions: (1) Translational attenuation that prevents excessive accumulation of unfolded proteins, (2) up-regulation of ER chaperones and folding enzymes, such as GRP78 and glucose-regulated protein 94 kD (GRP94) are involved in the general folding process to increase the protein folding capacity, (3) enhanced ERAD of unfolded proteins, which strengthen ERAD ability to clear unfolded proteins and send them to the cytoplasm for proteasome-mediated degradation, (4) induction of apoptosis, which happens when the unfolded protein in the ER is overwhelming and the adaptive mechanisms fail to compensate by the first three aforementioned approaches [
1,
2,
3].
The UPR plays an important role in the maintenance of proteostasis by reducing the nascent and misfolded proteins, which are produced under a variety of conditions in physiology and diseases [
4,
5,
6,
7,
8]. ER stress and activation of the UPR are associated with intestinal epithelial cell damage and apoptosis in CD [
5,
6,
7,
8]. UPR-associated genes (e.g., IRE1α, ATF6, and XBP1) have also been implicated in the genetic analysis of CD [
5,
6,
7,
8]. A number of studies have shown that ER stress and the UPR play a critical part in shaping immune cell differentiations and functions in order to mount either a protective or a destructive immune response in the host depending upon various conditions [
5,
8]. Furthermore, intestinal epithelial cells and microbiota contribute to the complexity and dynamic interaction with immune cells within the inflamed gut to resolve the tissue damage, which is induced by a variety of cytokines [
6,
7]. Therefore, it is understandable that the UPR with its downstream signaling pathways is required in the maintenance of intestinal homeostasis. Moreover, dysfunction of the ER stress response contributes to the pathogenesis of IBD and the complications, such as intestinal fibrosis [
9].
2. The Cause of the UPR
In physiological conditions, the ER is responsible for the entry and release of calcium, protein synthesis, and package, lipid metabolism [
10]. When cells are subjected to a wide range of stressful conditions, ER stress response reacts with generation of unfolded or misfolded proteins, which triggers UPR to rescue this cellular dysfunction. These stresses commonly include changes in calcium homeostasis, viral or bacterial infection, inflammation, nutrition or energy deficiency, hypoxia, lipid overload, altered redox status, as well as oncogene activation in cancer [
7,
10]. During UPR, transcription factors such as ATF6 and XBP-1 are activated and translocated to the nucleus to initiate transcription of genes, which are involved in inflammation, cell proliferation, and fibrosis [
9,
11]. In addition, ER stress plays a role in cellular differentiation, antigen presentation, and stem cell renewal capacity [
8]. In 1974, Drs. Claude, Duve, and Palade were awarded Nobel Prize in Physiology or Medicine “for their discoveries concerning the structural and functional organization of the cell", particularly related to the ER using an electronic microscope. ER stress has been studied in both physiologic and pathologic conditions in different organ systems [
3,
4,
5,
6,
7]. In the gastrointestinal system, it has been studied in epithelial cell differentiation and function, as well as CD during the process of intestinal epithelial cell damage [
5,
6,
7]. The role of ER stress in mesenchymal cells during the development of intestinal fibrosis has been recently reported and is discussed in detail in subsequent sections [
9].
4. The UPR in Macrophage and Mesenchymal Cells during the Immune Response
ER stress and immunity are usually intertwined together during different stages of the inflammatory process in a variety of human diseases [
5,
8].
In C/EBP homologous protein (CHOP)-/- mice, bleomycin-induced lung fibrosis was significantly attenuated compared to wild type mice [
28], while administration of tauroursodeoxycholic acid (TUDCA), a chemical chaperone, inhibited bleomycin-induced inflammation and fibrosis in mice [
28]. Endo et al. [
29] showed that LPS-induced inflammation in the lung of CHOP-/- mice was also attenuated, in addition to the decrease in neutrophil infiltration, IL-1β, and caspase-11 expression. However, Ayaub et al. [
30] showed that ECM deposition was increased with the proliferation of arginase-1-positive lung macrophages in CHOP-/- mice. Paradoxically, GRP78+/- haplo-insufficiency mice were significantly protected against bleomycin-induced lung fibrosis due to a decrease in the population of lung macrophages with positive stain for cleaved caspase-3. These data suggest that GRP78- and CHOP-mediated macrophage apoptosis may have opposite roles in response to bleomycin-induced fibrosis.
In a mouse model of nonalcoholic steatohepatitis, CHOP-/- mice demonstrated severe liver damage, inflammation, and fibrosis compared to CHOP wild type due to the increase in activated macrophages. Persistence of net accumulation of these activated macrophages in the liver potentiated liver steatohepatitis in CHOP-/- mice [
31]. In another study, Yao et al. [
32] reported that CHOP-/- diminished alternatively-activated-macrophage phenotype (M2) and reduced M2 filtration in the mouse lung after bleomycin treatment. Activated M2 macrophages secreted TGF-β and plate-derived-growth-factor (PDGF) to induce activation of myofibroblasts and led to tissue fibrosis. Taken together, the role of CHOP and GRP78 during ER stress should be examined and interpreted carefully because they may have opposite effects on macrophage activation and proliferation depending upon cell type, tissue, disease stage, and context.
During the dysregulated wound healing process, intestinal macrophage not only recruits surrounding mesenchymal cells, such as subepithelial myofibroblasts, to come into the inflamed area but also activates itself and subepithelial myofibroblasts [
33,
34,
35]. Once activated, these cells release a variety of inflammatory cytokines and overproduce extracellular matrix proteins. Finally, these events thicken the tissue layer, destroy the regular motility function and the capability of nutrition absorption in the gut [
33,
34,
35].
Macrophages are essential immune cells for the maintenance of tissue homeostasis in the intestinal mucosa barrier. They are actively involved in the repairing process of wound healing, particularly in the context of intestinal damage and tissue repair in IBD [
36]. Phenotypic plasticity of macrophages from classical M1 to alternative M2 is controlled by a variety of cytokines, such as IFN-γ and IL-4. The IL-4-derived M2 can further differentiate into activated myofibroblasts [
36]. Alpha-smooth muscle actin (α-SMA) positive-myofibroblasts are central to the wound healing process and highly expressed in patients with fibrostenotic CD [
37,
38]. They contribute to fibrosis by producing excessive amounts of ECM proteins [
33,
34].
Our recent study showed that the CD38+/M1 MΦ decreased and CD163+/M2 MΦ increased significantly in macrophages, which were isolated from the colon of 2, 4, 6-Trinitrobenzenesulfonic acid (TNBS)-treated mice compared to ethanol-treated mice [
39]. The M2 MΦ was increased in the colon of TNBS treated mice due to MΦ-to-myofibroblast transition, where M1 MΦ decreased significantly. Treatment with tunicamycin significantly increased the ER stress marker, GRP78, and CD163+/M2 MΦ population. Treatment with IL-4 had a similar effect on the numbers of CD163+/M2 MΦ. Treatment with a green tea compound, epigallocatechin-3-gallate (EGCG), an ER stress inhibitor, suppressed IL-4-induced increase in CD163+/M2 MΦ. The effect was blocked with a neutralizing antibody against the 67-kDa laminin receptor (67LR), a reported EGCG-binding receptor. The inhibitory effect of EGCG was associated with an increase in 67LR+/vimentin+ macrophages isolated from mice with TNBS-induced colitis compared to the ethanol-treated group. EGCG also suppressed the tunicamycin-induced increase in GRP78 and production of α-SMA+ macrophages during MΦ-to-myofibroblast transition through its binding to 67LR [
39]. These data suggest that ER stress may regulate the phenotypic change of macrophages and macrophage-to-myofibroblast transition. However, the exact role of macrophages during the development of intestinal fibrosis in patients with CD still awaits further study.
5. Epigenetic Regulation of the UPR
The rapidly developing field of epigenetics demonstrates the great potential to elucidate the pathological mechanism of abnormal gene expression due to the changes in the structure and function of the chromatin. These changes can be caused by environmental factors, such as hypoxia, microbial toxins (e.g., Shiga toxigenic factors that degrade GRP78), and dietary factors (e.g., iron) [
40,
41].
Epigenetic mechanisms affect gene expression and cellular function through three distinctive but also interconnected mechanisms: (1) Chromatin structure modulation, (2) DNA methylation, and (3) RNA interference by small non-coding RNAs, i.e., microRNAs [
42,
43,
44,
45].
Llinàs-Arias et al. [
46] showed that the small p97/VCP-interacting protein (SVIP), an endogenous inhibitor of ERAD, underwent DNA hypermethylation-associated silencing in high-risk patients who manifest poor clinical outcomes. The dependence of SVIP-hypermethylated cancer cells on aerobic glycolysis and glucose was also related to the sensitivity to an inhibitor of the glucose transporter GLUT1. This study demonstrated that how epigenetics affects ER stress and how SVIP epigenetic silencing in cancer may be applicable to the therapy that targets glucose transporters. Little is known about GRP78 proteostasis and the role of its posttranslational modifications in ER stress. Sieber et al. [
47] reported a novel proteostatic mechanism that is dependent on the posttranslational modification of GRP78, allowing cells to differentially regulate protein production during ER stress. ER stress led to de novo biosynthesis of non-trimethylated GRP78, whereas homeostatic, N-lysine methyltransferase 21A (METTL21A)-dependent lysine 585–trimethylated GRP78 was reduced. In other words, ER stress triggered the de novo synthesis of non-trimethylated GRP78 and simultaneous degradation of existing, lysine-trimethylated GRP78. This previously unrecognized mechanism suggests the lack of posttranslational modification may alter the conformation of GRP78 in a way that may be beneficial during ER stress to secure cell survival.
The emergence of miRNAs during the course of UPR-mediated adaptive and apoptotic signaling has provided more mechanistic understanding of their roles in gene regulation in vivo. For example, miR-379 targets (and therefore represses) Edem3, which encodes an inhibitor of ER stress, whereas miR-494, another miRNA in the miR-379 cluster, targets Atf3, a repressor of CHOP [
48].
Differential microRNAs’ activities contribute to pro-adaptive/survival and pro-apoptotic UPR signaling by targeting the three main sensors of ER stress, including IRE1, PERK, and ATF6, in vitro and in vivo [
49,
50]. For example, Upton et al. [
51] reported that IRE1α RNase activation caused selective microRNAs (miRs -17, -34a, -96, and -125b) degradation that normally represses translation of Caspase-2 mRNA, leading to activation of the mitochondrial apoptotic pathway.
Moreover, our recent study showed that the UPR and its downstream signaling pathways could be manipulated through epigenetic regulations [
9]. We showed that expression of ER stress sensors increased significantly in subepithelial myofibroblasts of strictured intestine from patients with fibrostenotic CD [
9]. The increase in the ER stress response featured with overexpression of GRP78, XBP1s, and ATF6α can be reproduced in the normal subepithelial myofibroblasts when treated with tunicamycin, which is an ER stress agonist [
9]. The increased levels of ER stress in affected ileum was associated with silencing of miR-199a-5p by DNA-methyltransferase 1 (DNMT1)-mediated promoter hypermethylation [
9]. At rest condition, miR-199a-5p targeted ER stressors including GRP78, ATF6, and XBP1s for their degradation [
9]. Restoration of miR-199a-5p through a DNA methylation inhibitor, 5-azacytidine, via inhibition of DNMT1 function, suppressed ER stress-induced myofibroblasts activation and excess ECM production [
9]. During ER stress, DNMT1 upregulated and led to hypermethylation of miR-199a-5p and its silencing. This silencing in miR-199a-5p led to the loss of its inhibition on the ER components and causes upregulation of the ER stress components, TGF-β1 levels, and resultant fibrosis [
9]. Put together, this epigenetic evidence will improve our understanding of the molecular mechanism of fibrosis within the context of ER stress and the UPR ().
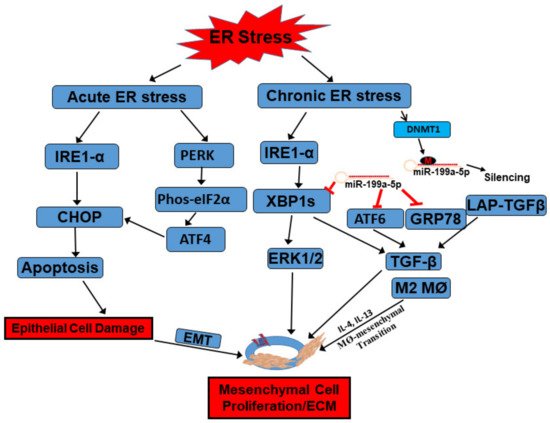
Figure 1. Functions of the UPR in the development of intestinal fibrosis in Crohn’s disease. When intestinal epithelial cells (IECs) are subject to acute ER stress, ER stress sensors IRE1α and PERK can be activated with detached association from binding partner GRP78. Downstream signaling, including CHOP and eIF2α/ATF4, are further activated to induce apoptosis in IECs. Meanwhile, inflammatory cytokines, such as IL-4 and IL-13, induced activation of macrophages (M2) as well as macrophage-to-mesenchymal transition. TGF-β can be secreted from this transition to further activate mesenchymal cells, such as subepithelial myofibroblasts, to proliferate and induce extracellular matrix protein production. When the intestine is subject to chronic inflammation-induced ER stress, IRE1α catalyzes non-canonical splicing of X-box binding protein 1 (XBP1) mRNA into the constitutively active form XBP1s, which activates ERK1/2 to stimulate mesenchymal cell proliferation. The increased UPR is also associated with increased silencing of miR-199a-5p by DNMT1-mediated promoter hypermethylation. At rest condition, miR-199a-5p targets different ER stressors, including GRP78, ATF6, and XBP1s, through complementary binding to their promoter regions for their degradation. ATF6 and XBP1 both serve as transcription factors and activate ER stress-induced myofibroblasts activation through upregulation of TGF-β and excess ECM production. GRP78 can bind to latent associated peptide (LAP)-TGFβ to activate TGF-β. All these factors can finally contribute to the development of intestinal fibrosis. Refer to context for details.
In summary, epigenetic regulation of ER stress and the UPR may provide a deeper understanding of how a variety of UPR branches and downstream signaling pathways contribute to the pathogenesis of different diseases, suggesting novel pharmacological targets of the ER stress components.