Aging is a fundamental biological process accompanied by a general decline in tissue function. Indeed, as the lifespan increases, age-related dysfunction, such as cognitive impairment or dementia, will become a growing public health issue.
- AMPK
- SIRT1
- mTOR
- aging
- senescence
- health span
- intervention
- signaling
1. Introduction
Nowadays, the population and life expectancy of humans are significantly increasing, due in large part to improvements in nutrients, medicine and environments, etc., which results in a decrease in mortality from age-related diseases, such as heart disease, cancer and stroke [1]. The world’s total population was about 6.76 billion in 2008, and the number increased to 7.59 billion in 2018. Sixteen percent will be over the age of 65 by 2050, up from 9% in 2019 in the world, suggesting that the age group of 65 and over is growing the fastest. Aging is a fundamental biological process accompanied by a general decline in tissue function and increased risk for many age-related diseases. According to the National Council on Aging, about 92 percent of the elderly have at least one age-related disease and 77% have at least two. Heart disease, ischemic stroke, cancer and diabetes are among the most common disorders. For example, as the lifespan increases, cognitive impairment or dementia will become a growing public health issue. According to current estimates, almost 36 million people have dementia worldwide, and this number is expected to reach 66 million by 2030 and 115 million by 2050 [2]. The population with dementia is expected to increase to a total of over 13 million in the United States alone. The most well-known form of dementia is Alzheimer’s disease (AD), but a large percentage of aged cognitively impaired persons are not due to AD but, rather, the normal aging process. The vast majority of older adults suffer declines in cognitive functions, interfering with their ability to participate and engage in meaningful activities [3]. In addition to AD, deterioration in fine motor control, gait and balance are among the most important health problems in the elderly. Now, falls are the leading cause of injury-related death and the third-leading cause of poor health among persons aged 65 years and older [4]. In 2013, the cost of these injuries caused by falls was US $34 billion. Thus, there is a critical need in understanding underlying cellular and molecular mechanisms regulating aging, which will allow us to modify the aging process for healthy aging and alleviate age-related disease.
2. AMPK Signaling
AMPK (a serine/threonine protein kinase) is an essential energy sensor engaged in modulating our whole-body level of metabolic energy balance [5][6]. In mammals, AMPK comprises of two isoforms of the α and β subunits each and three isoforms of the γ subunits to form a combination of 12 different αβγ isoforms. The α subunit encodes an N-terminal protein kinase domain, which interacts with a C-terminal regulatory domain via its well-known activation segment threonine 172 (T172) to mediate AMPK’s catalytic activity. The β subunit has a carbohydrate-binding module (CBM), and its C-terminal domain connects the γ subunit and the C-terminal domain of the α subunit. The γ subunit holds four cystathionine-β-synthase (CBS) regions referring to adenine nucleotide binding, which ensures the monitoring and regulation for AMP/ATP ratio [7][8][9]. Both the β (β1 and β2) and γ (γ1, γ2 and γ3) subunits regulate AMPK’s phosphorylation and activity. Traditionally, AMPK signaling is one of the central regulators of cellular and organismal metabolism in eukaryotes, playing important roles in regulating growth and reprogramming metabolism. Recently, AMPK signaling has been connected to aging and the lifespan, as AMPK can control the regulation of cellular homeostasis, resistance to stress, cell survival and growth, cell death and autophagy. Supportively, specific AMPK activation protects against aging and extends the lifespan in Caenorhabditis elegans (C. elegans) and rodents.
3. Sirtuin Signaling
Sirtuin (SIRT) was first discovered in the 1970s in nature and is an essential factor in delaying cellular senescence and extending organismal lifespan [10]. SIRT1 is the most well-studied among the seven SIRT isoforms in humans [11]. SIRT1 is an NAD+-dependent deacetylase and, thus, can deacetylate tumor suppressor p53 protein [12][13], the DNA repair factor Ku70 [14], NF-κB [15], the signal transducer and activator of transcription 3 (STAT3) [16] and the FOXO family of forkhead transcription factors (Figure 1). The roles of Sirtuin on the suppression of cellular senescence is primarily mediated through delaying age-related telomere attrition, sustaining genome integrity and promoting DNA damage repair. SIRT1 can improve the ability to induce cell cycle arrest and oxidative stress resistance and inhibits cell death [17] and apoptotic pathways [18]. It has been verified that Ku70 blocks the death of stress-induced apoptotic cells by sequestering the proapoptotic factor Bax away from the mitochondria. NF-κB is involved in upregulating gene products controlling cell survival. SIRT1 participates in many age-related processes and disorders, such as neurodegenerative diseases and cardiovascular diseases, etc. [19].
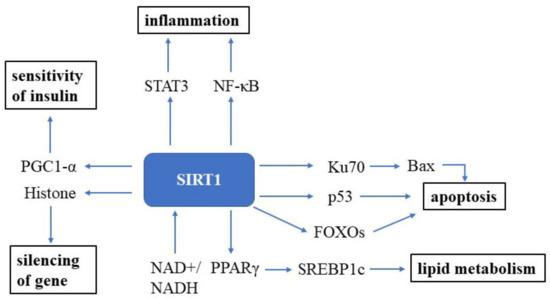
Figure 1. SIRT1 signaling in the cell. SIRT1 is a well-known NAD+-dependent deacetylase that impacts several molecules to promote health. Abbreviations: STAT3: signal transducer and activator of transcription 3; NF-κB: NF-kappa B; Bax: bcl-2-associated X protein; FOXOs: forkhead box transcription factors; PPARγ: peroxisome proliferator-activated receptors; NAD+: oxidized nicotine adenine dinucleotide; NADH: reduced nicotine adenine dinucleotide.
4. MTOR Signaling
The mammalian target of rapamycin (mTOR) is a serine/threonine kinase, which is a part of the phosphoinositide 3-kinase (PI3K)-related kinase family. mTOR functions as an intracellular energy sensor and a central regulator of growth, proliferation, metabolism and aging [20][21][22]. mTOR exists as two distinct protein complexes, mTOR complex 1 (mTORC1) and 2 (mTORC2) [20]. mTORC1 consists of five parts, including mTOR, the regulatory-associated protein of mTOR (Raptor), mammalian lethal with Sec13 protein 8 (mLST8), proline-rich Akt substrate 40 kDa (PRAS40) and DEP-domain-containing mTOR-interacting protein (DEPTOR). mTORC2 contains six elements, including mTOR, rapamycin-insensitive companion of mTOR (Rictor), mammalian stress-activated protein (mSIN1), protein observed with Rictor-1 (Protor-1), mLST8 and DEPTOR. DEPTOR is negatively regulated by both mTORC1 and mTORC2. Inhibiting DEPTOR will activate ribosomal protein S6 kinase (S6K), Akt and SGK1, resulting in cell growth and proliferation (Figure 2) [23].
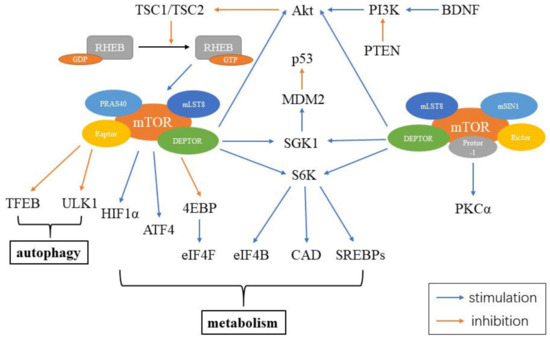
Figure 2. Constituents of mTORC and mTOR signaling in the cell. mTORC1 includes mTOR, Raptor, mLST8, PRAS40 and DEPTOR, while mTORC2 contains mTOR, Rictor, mSIN1, Protor-1, mLST8 and DEPTOR. Abbreviations: RHEB: ras homolog enriched in brain; GDP: guanosine diphosphate; GTP: guanosine triphosphate; PRAS40: proline-rich Akt substrate 40 kDa; mLST8: mammalian lethal with Sec13 protein 8; Raptor: the regulatory-associated protein of mTOR; DEPTOR: DEP-domain-containing mTOR-interacting protein; TFEB: transcription factor EB; HIF1α: hypoxia inducible factor 1α; ATF4: activating transcription factors 4; 4EBP: 4E binding protein; eIF4F: eukaryotic initiation factor 4F; eIF4B: eukaryotic initiation factor 4B; CAD: carbamoyl-phosphate synthetase; S6K: S6 kinase; SGK1: glucocorticoid induced protein kinase 1; MDM2: murine double minute 2; Akt: protein kinase B; PI3K: phosphoinositide 3-kinase; PTEN: phosphatase and tensin homolog; BDNF: brain-derived neurotrophic factor; mSIN1: mammalian stress-activated protein; Protor-1: protein observed with Rictor-1; Rictor: rapamycin-insensitive companion of mTOR; PKCα: protein kinase Cα.
5. Interplay between the AMPK, SIRT1 and mTOR Signaling Pathways
AMPK, SIRT1 and mTOR can also interact with each other, forming an intertwined web. For example, AMPK and SIRT1 increase the expression of Atgs by upregulating FOXOs and PGC-1α and downregulating mTORC1 [24]. Wang et al. showed that the antiaging effects of resveratrol in zebrafish retina involved the activation of the AMPK, SIRT1 and mTOR signaling pathways [25].
5.1. Crosstalk between AMPK and SIRT1 Signaling
SIRT1 activates AMPK by stimulating LKB1 via deacetylation, while AMPK activates SIRT1 by enhancing the NAD+ level [26]. Kim et al. found that resveratrol ameliorated oxidative stress and mitochondrial dysfunction by activating the SIRT1/AMPK/PGC-1α axis in an age-related renal injury [27]. Six weeks of exercise, resveratrol and the combination of both significantly improved the expression of p-AMPK and SIRT1 in the brains of aged rats to exert protective effects [28]. Swimming modulated the expression levels of SIRT1/PGC1α, AMPK and FOXO3a in the gastrocnemius muscles of 3-, 12- and 18-month-old rats and inhibited aged hippocampus cell apoptosis and inflammation through IGF1/PI3K/Akt signaling and AMPK/SIRT1/PGC1α signaling [29][30]. Regular aerobic exercise can balance apoptosis and autophagy within the corpus striatum in aged rats through the AMPK/SIRT1 pathway [31]. Wheel running can also activate lysosomal and autophagic functions in the mouse brain via AMPK/SIRT1/TFEB [32]. In SAMP8 mice, a model for aging, wheel running influenced the mitochondrial function via modulating the SIRT1/AMPK pathways [33]. E6155 reduced the fasting glucose and improved the tolerance to oral glucose and insulin through the SIRT1/LKB1/AMPK axis [34]. A moderate dose of resveratrol improved the mitochondrial biogenesis and function via AMPK stimulation induced by SIRT1 [26]. The AMPK/SIRT1/FOXO1 axis also participated in modulating apoptosis in bovine intracellular adipocytes [35]. Glucose restriction regulated nicotinamide phosphoribosyltransferase (NAMPT), an important rate-limiting enzyme in the synthesis of NMN, to stimulate SIRT1 with the help of AMPK to have a negative effect on skeletal myoblast differentiation [36].
5.2. Interaction between AMPK and mTOR Signaling
AMPK interacts with mTOR in two main ways, by directly phosphorylating Raptor and indirectly phosphorylating TSC2, resulting in the activation of the GTPase-activating protein and the combination of Rheb on lysosomal membranes [37][38]. A moderate dose of bilberry anthocyanin (MBA) upregulated the expression of OCLN, ZO-1 and autophagy associated-proteins ATP6 V0C, ATG4D and CTSB to induce autophagy through the AMPK/mTOR pathway and then improved the intestinal epithelial barrier function and oxidative stress resistance effects in aging female rats [39]. Genistein dose- and time-dependently activated LKB1/AMPK/mTOR signaling to induce autophagy, which protected VSMCs from aging [40]. Acarbose competitively and reversibly inhibited salivary and pancreatic α-amylases and small intestine brush border α-glucosidases, which then decreased postprandial glucose and subsequently modulated the AMPK- and mTORC1-regulated metabolism pathways, leading to the improved survival of Apc mice, a mimic of familial adenomatous polyposis in humans [41]. Metformin improves cartilage degeneration in osteoarthritic mice through the AMPK/mTOR pathway [42]. Qing et al. found that metformin regulated the AMPK/mTOR/NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome signaling pathways to induce M2 macrophage polarization, leading to the promotion of wound healing [43]. In osteoarthritic mice, a local intra-articular injection of resveratrol induced autophagy through AMPK/mTOR signaling and attenuated cartilage degeneration [44]. Exogenous hydrogen sulfide (H2S) rescued cardioprotection from ischemic postconditioning by stimulating autophagy via the AMPK/mTOR pathway in isolated aged rat hearts and aged cardiomyocytes [45]. Oleuropein aglycone (OLE), the main polyphenol in extra virgin olive oil, exerted beneficial health effects by activating autophagy through the AMPK/mTOR pathways in cultured neuroblastoma cells and OLE-fed mice [46]. CRMs ameliorated hyperglycemia-induced senescence and epithelial–mesenchymal transition (EMT) by activating AMPK/mTOR signaling [47]. In ApoE-deficient mice, CR upregulated Fgf21 to stimulate AMPK/mTOR signaling, leading to a decrease in the formation of neurofibrillary tangles by inhibiting tau phosphorylation [48].
5.3. Interplay between SIRT1 and mTOR Signaling
The relationship between SIRT1 and mTOR remains largely unexplored. It was found that SIRT1 inhibits mTOR signaling by interacting with TSC2 [49]. SIRT1 was needed for rapamycin to have an effect on high-glucose-induced mesangial cells senescence [50]. The mTORC1 inhibitor rapamycin could enhance the expression levels of SIRT1 and AMPK and improve the deacetylase ability of SIRT1 in AML12 hepatocytes [51]. mTOR and SIRT1 cooperated to promote the proliferation of intestinal stem cells in CR [52]. SIRT1 decreased the autophagy and mitochondrial function in embryonic stem cells by downregulating the mTOR pathways in response to oxidative stress [53]. Similarly, in alcoholic liver disease mice and patients, the lack of DEPTOR and SIRT1 induced the abnormal activation of mTORC1, resulting in an increased phosphorylation level of mTOR and S6K1 to aggravate inflammation and acute-on-chronic liver injuries [54].
This entry is adapted from the peer-reviewed paper 10.3390/cells10030660
References
- Zhao, R.; Stambler, I. The Urgent Need for International Action for Anti-aging and Disease Prevention. Aging Dis. 2020, 11, 212–215.
- Brayne, C.; Miller, B. Dementia and aging populations-A global priority for contextualized research and health policy. PLoS Med. 2017, 14, e1002275.
- Harada, C.; Natelson Love, M.; Triebel, K. Normal cognitive aging. Clin. Geriatr. Med. 2013, 29, 737–752.
- Evitt, C.; Quigley, P. Fear of falling in older adults: A guide to its prevalence, risk factors, and consequences. Rehabil. Nurs. Off. J. Assoc. Rehabil. Nurses 2004, 29, 207–210.
- Novelle, M.; Ali, A.; Diéguez, C.; Bernier, M.; de Cabo, R. Metformin: A Hopeful Promise in Aging Research. Cold Spring Harb. Perspect. Med. 2016, 6, a025932.
- Wang, B.; Yang, J.; Zhang, H.; Smith, C.; Jin, K. AMPK Signaling Regulates the Age-Related Decline of Hippocampal Neurogenesis. Aging Dis. 2019, 10, 1058–1074.
- Carling, D. AMPK signalling in health and disease. Curr. Opin. Cell Biol. 2017, 45, 31–37.
- Morgunova, G.V.; Klebanov, A.A. Age-related AMP-activated protein kinase alterations: From cellular energetics to longevity. Cell Biochem. Funct. 2019, 37, 169–176.
- Tamás, P.; Hawley, S.; Clarke, R.; Mustard, K.; Green, K.; Hardie, D.; Cantrell, D. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J. Exp. Med. 2006, 203, 1665–1670.
- Lee, S.; Lee, J.; Lee, H.; Min, K. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34.
- Wang, Y.; He, J.; Liao, M.; Hu, M.; Li, W.; Ouyang, H.; Wang, X.; Ye, T.; Zhang, Y.; Ouyang, L. An overview of Sirtuins as potential therapeutic target: Structure, function and modulators. Eur. J. Med. Chem. 2019, 161, 48–77.
- Qureshi, A.; Ali, Z.; Suri, M.; Kim, S.; Shatla, A.; Ringer, A.; Lopes, D.; Guterman, L.; Hopkins, L. Intra-arterial third-generation recombinant tissue plasminogen activator (reteplase) for acute ischemic stroke. Neurosurgery 2001, 49, 41–48, discussion 48–50.
- Broderick, J.; Lu, M.; Jackson, C.; Pancioli, A.; Tilley, B.; Fagan, S.; Kothari, R.; Levine, S.; Marler, J.; Lyden, P.; et al. Apolipoprotein E phenotype and the efficacy of intravenous tissue plasminogen activator in acute ischemic stroke. Ann. Neurol. 2001, 49, 736–744.
- Cohen, H.Y.; Miller, C.; Bitterman, K.J.; Wall, N.R.; Hekking, B.; Kessler, B.; Howitz, K.T.; Gorospe, M.; De Cabo, R.; Sinclair, D.A. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 2004, 305, 390–392.
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380.
- Bernier, M.; Paul, R.K.; Martin-Montalvo, A.; Scheibye-Knudsen, M.; Song, S.; He, H.-J.; Armour, S.M.; Hubbard, B.P.; Bohr, V.A.; Wang, L.; et al. Negative regulation of STAT3 protein-mediated cellular respiration by SIRT1 protein. J. Biol. Chem. 2011, 286, 19270–19279.
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015.
- Liu, Y.; Weng, W.; Gao, R.; Liu, Y. New Insights for Cellular and Molecular Mechanisms of Aging and Aging-Related Diseases: Herbal Medicine as Potential Therapeutic Approach. Oxidative Med. Cell. Longev. 2019, 2019, 4598167.
- Zhao, L.; Cao, J.; Hu, K.; He, X.; Yun, D.; Tong, T.; Han, L. Sirtuins and their Biological Relevance in Aging and Age-Related Diseases. Aging Dis. 2020, 11, 927–945.
- Albert, V.; Hall, M.N. mTOR signaling in cellular and organismal energetics. Curr. Opin. Cell Biol. 2015, 33, 55–66.
- Di Francesco, A.; Diaz-Ruiz, A.; De Cabo, R.; Bernier, M. Intermittent mTOR Inhibition Reverses Kidney Aging in Old Rats. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2018, 73, 843–844.
- Apelo, S.I.A.; Lamming, D.W. Rapamycin: An InhibiTOR of Aging Emerges from the Soil of Easter Island. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2016, 71, 841–849.
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886.
- Escobar, K.; Cole, N.; Mermier, C.; Van Dusseldorp, T. Autophagy and aging: Maintaining the proteome through exercise and caloric restriction. Aging Cell 2019, 18, e12876.
- Wang, N.; Luo, Z.; Jin, M.; Sheng, W.; Wang, H.; Long, X.; Wu, Y.; Hu, P.; Xu, H.; Zhang, X. Exploration of age-related mitochondrial dysfunction and the anti-aging effects of resveratrol in zebrafish retina. Aging 2019, 11, 3117–3137.
- Price, N.; Gomes, A.; Ling, A.; Duarte, F.; Martin-Montalvo, A.; North, B.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690.
- Kim, E.; Lim, J.; Kim, M.; Ban, T.; Jang, I.; Yoon, H.; Park, C.; Chang, Y.; Choi, B. Resveratrol, an Nrf2 activator, ameliorates aging-related progressive renal injury. Aging 2018, 10, 83–99.
- Liao, Z.; Chen, J.; Xiao, M.; Sun, Y.; Zhao, Y.; Pu, D.; Lv, A.; Wang, M.; Zhou, J.; Zhu, S.; et al. The effect of exercise, resveratrol or their combination on Sarcopenia in aged rats via regulation of AMPK/Sirt1 pathway. Exp. Gerontol. 2017, 98, 177–183.
- Lin, J.; Kuo, W.; Baskaran, R.; Kuo, C.; Chen, Y.; Chen, W.; Ho, T.; Day, C.; Mahalakshmi, B.; Huang, C. Swimming exercise stimulates IGF1/ PI3K/Akt and AMPK/SIRT1/PGC1α survival signaling to suppress apoptosis and inflammation in aging hippocampus. Aging 2020, 12, 6852–6864.
- Huang, C.; Wang, T.; Tung, Y.; Lin, W. Effect of Exercise Training on Skeletal Muscle SIRT1 and PGC-1α Expression Levels in Rats of Different Age. Int. J. Med. Sci. 2016, 13, 260–270.
- Liu, W.; Wang, Z.; Xia, Y.; Kuang, H.; Liu, S.; Li, L.; Tang, C.; Yin, D. The balance of apoptosis and autophagy via regulation of the AMPK signal pathway in aging rat striatum during regular aerobic exercise. Exp. Gerontol. 2019, 124, 110647.
- Huang, J.; Wang, X.; Zhu, Y.; Li, Z.; Zhu, Y.; Wu, J.; Qin, Z.; Xiang, M.; Lin, F. Exercise activates lysosomal function in the brain through AMPK-SIRT1-TFEB pathway. CNS Neurosci. Ther. 2019, 25, 796–807.
- Bayod, S.; Guzmán-Brambila, C.; Sanchez-Roige, S.; Lalanza, J.; Kaliman, P.; Ortuño-Sahagun, D.; Escorihuela, R.; Pallàs, M. Voluntary exercise promotes beneficial anti-aging mechanisms in SAMP8 female brain. J. Mol. Neurosci. MN 2015, 55, 525–532.
- Liu, P.; Feng, T.; Zuo, X.; Wang, X.; Luo, J.; Li, N.; Han, X.; Zhu, N.; Xu, S.; Xu, Y.; et al. A novel SIRT1 activator E6155 improves insulin sensitivity in type 2 diabetic KKA mice. Biochem. Biophys. Res. Commun. 2018, 498, 633–639.
- Liu, X.; Zhao, H.; Jin, Q.; You, W.; Cheng, H.; Liu, Y.; Song, E.; Liu, G.; Tan, X.; Zhang, X.; et al. Resveratrol induces apoptosis and inhibits adipogenesis by stimulating the SIRT1-AMPKα-FOXO1 signalling pathway in bovine intramuscular adipocytes. Mol. Cell. Biochem. 2018, 439, 213–223.
- Fulco, M.; Cen, Y.; Zhao, P.; Hoffman, E.P.; McBurney, M.W.; Sauve, A.A.; Sartorelli, V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 2008, 14, 661–673.
- Jung, C.; Ro, S.; Cao, J.; Otto, N.; Kim, D. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295.
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226.
- Li, J.; Zhao, R.; Zhao, H.; Chen, G.; Jiang, Y.; Lyu, X.; Wu, T. Reduction of Aging-Induced Oxidative Stress and Activation of Autophagy by Bilberry Anthocyanin Supplementation via the AMPK-mTOR Signaling Pathway in Aged Female Rats. J. Agric. Food Chem. 2019, 67, 7832–7843.
- Lee, K.; Kim, J.; Choi, H. Genistein-induced LKB1-AMPK activation inhibits senescence of VSMC through autophagy induction. Vasc. Pharmacol. 2016, 81, 75–82.
- Dodds, S.G.; Parihar, M.; Javors, M.; Nie, J.; Musi, N.; Sharp, Z.D.; Hasty, P. Acarbose improved survival for Apc mice. Aging Cell 2020, e13088.
- Feng, X.; Pan, J.; Li, J.; Zeng, C.; Qi, W.; Shao, Y.; Liu, X.; Liu, L.; Xiao, G.; Zhang, H.; et al. Metformin attenuates cartilage degeneration in an experimental osteoarthritis model by regulating AMPK/mTOR. Aging 2020, 12, 1087–1103.
- Qing, L.; Fu, J.; Wu, P.; Zhou, Z.; Yu, F.; Tang, J. Metformin induces the M2 macrophage polarization to accelerate the wound healing via regulating AMPK/mTOR/NLRP3 inflammasome singling pathway. Am. J. Transl. Res. 2019, 11, 655–668.
- Qin, N.; Wei, L.; Li, W.; Yang, W.; Cai, L.; Qian, Z.; Wu, S. Local intra-articular injection of resveratrol delays cartilage degeneration in C57BL/6 mice by inducing autophagy via AMPK/mTOR pathway. J. Pharmacol. Sci. 2017, 134, 166–174.
- Chen, J.; Gao, J.; Sun, W.; Li, L.; Wang, Y.; Bai, S.; Li, X.; Wang, R.; Wu, L.; Li, H.; et al. Involvement of exogenous H2S in recovery of cardioprotection from ischemic post-conditioning via increase of autophagy in the aged hearts. Int. J. Cardiol. 2016, 220, 681–692.
- Rigacci, S.; Miceli, C.; Nediani, C.; Berti, A.; Cascella, R.; Pantano, D.; Nardiello, P.; Luccarini, I.; Casamenti, F.; Stefani, M. Oleuropein aglycone induces autophagy via the AMPK/mTOR signalling pathway: A mechanistic insight. Oncotarget 2015, 6, 35344–35357.
- Dong, D.; Cai, G.; Ning, Y.; Wang, J.; Lv, Y.; Hong, Q.; Cui, S.; Fu, B.; Guo, Y.; Chen, X. Alleviation of senescence and epithelial-mesenchymal transition in aging kidney by short-term caloric restriction and caloric restriction mimetics via modulation of AMPK/mTOR signaling. Oncotarget 2017, 8, 16109–16121.
- Rühlmann, C.; Wölk, T.; Blümel, T.; Stahn, L.; Vollmar, B.; Kuhla, A. ApoELong-term caloric restriction in -deficient mice results in neuroprotection via Fgf21-induced AMPK/mTOR pathway. Aging 2016, 8, 2777–2789.
- Ghosh, H.S.; McBurney, M.; Robbins, P.D. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS ONE 2010, 5, e9199.
- Zhang, S.; Cai, G.; Fu, B.; Feng, Z.; Ding, R.; Bai, X.; Liu, W.; Zhuo, L.; Sun, L.; Liu, F.; et al. SIRT1 is required for the effects of rapamycin on high glucose-inducing mesangial cells senescence. Mech. Ageing Dev. 2012, 133, 387–400.
- Wang, Y.; Li, X.; He, Z.; Chen, W.; Lu, J. Rapamycin attenuates palmitate-induced lipid aggregation by up-regulating sirt-1 signaling in AML12 hepatocytes. Pharmazie 2016, 71, 733–737.
- Igarashi, M.; Guarente, L. mTORC1 and SIRT1 Cooperate to Foster Expansion of Gut Adult Stem Cells during Calorie Restriction. Cell 2016, 166, 436–450.
- Ou, X.; Lee, M.R.; Huang, X.; Messina-Graham, S.; Broxmeyer, H.E. SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells 2014, 32, 1183–1194.
- Chen, H.; Shen, F.; Sherban, A.; Nocon, A.; Li, Y.; Wang, H.; Xu, M.J.; Rui, X.; Han, J.; Jiang, B.; et al. DEP domain-containing mTOR-interacting protein suppresses lipogenesis and ameliorates hepatic steatosis and acute-on-chronic liver injury in alcoholic liver disease. Hepatology 2018, 68, 496–514.