Macrophages within solid tumors and metastatic sites are heterogenous populations with different developmental origins and substantially contribute to tumor progression. A number of tumor-promoting phenotypes associated with both tumor- and metastasis-associated macrophages are similar to innate programs of embryonic-derived tissue-resident macrophages. In contrast to recruited macrophages originating from marrow precursors, tissue-resident macrophages are seeded before birth and function to coordinate tissue remodeling and maintain tissue integrity and homeostasis. Both recruited and tissue-resident macrophage populations contribute to tumor growth and metastasis and are important mediators of resistance to chemotherapy, radiation therapy, and immune checkpoint blockade. Thus, targeting various macrophage populations and their tumor-promoting phenotypes holds therapeutic promise.
- tumor-associated macrophages
- tissue-resident macrophages
- tumor microenvironment
- monocyte
- metastasis-associated macrophage
- trained immunity
- depletion
- recruitment
- repolarization
- cancer
- immune therapy
1. Introduction
The tumor microenvironment (TME) is a diverse niche in which tumor, stromal, and immune cells dynamically interact via secreted factors and physical engagement, all within a dynamic extracellular matrix (ECM) [1]. Though the composition of the TME varies between tumor types, macrophages are often abundant and play a key role in tumor progression through promotion of tumor survival pathways and suppression of cytotoxic T cell responses [2]. Perhaps unsurprisingly, tumor cells co-opt a number of known tissue regulatory programs orchestrated by macrophage populations, including regulation of ECM, cell motility, chemotaxis, angiogenesis, and immune signaling pathways, to increase their survival [3]. Targeting macrophages and their tumor promotional programs is a major area of research in the pursuit of successful therapy [4]. It is also increasingly clear that targeting macrophages may be a beneficial approach to enhance the efficacy of conventional cytotoxic therapies and immune checkpoint blockade (ICB) [5]. Encouraging results from pre-clinical murine models are now being translated to the clinic with a number of clinical trials currently under investigation [6].
2. Targeting Macrophages to Potentiate Anti-Cancer Therapy
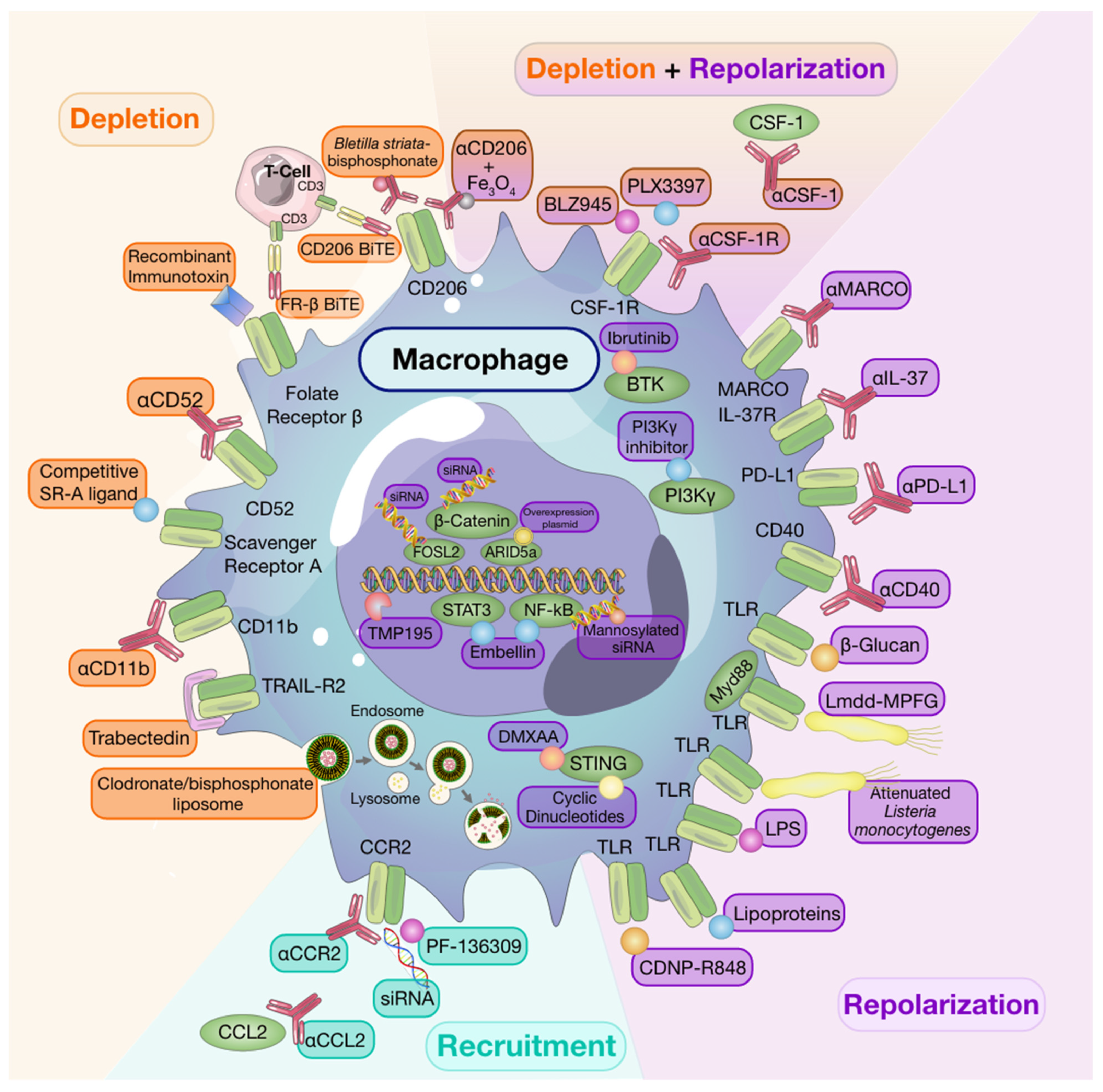
2.1. Macrophage Depletion
While targeted depletion of macrophages as monotherapy in preclinical models of established solid tumors is beneficial in some instances [8][9][10], other studies have revealed that, when used as a single agent, macrophage targeting strategies are minimally effective at slowing growth in established tumors [11]. It is interesting to note that for some studies wherein monotherapy was advantageous, depletion strategies targeted resident macrophage populations in addition to TAMs [9]. Despite limited evidence of efficacy for single-agent therapies to deplete macrophages in solid tumors, a number of clinical trials investigating macrophage depleting agents have been undertaken [12] and have also shown limited efficacy. However, it is worth noting that macrophage depletion following tumor resection in a spontaneous mouse model of melanoma reduced recurrence and metastasis [13], thus, highlighting timing of therapy as a consideration to achieve maximal therapeutic success [11].
Early monocyte and macrophage depletion methods relied on the use of bisphosphonate-packed and clodronate-loaded liposomes [14][15][16]. Clodronate-mediated TAM ablation demonstrated the crucial role of macrophages in orchestrating early tumor formation and progression in a mouse model of mammary cancer [17]. Depleting TAMs using clodronate-loaded liposomes was also shown to augment the anti-tumor effects of the protein kinase inhibitor, sorafenib, on tumor angiogenesis, growth, and metastasis in a hepatocellular carcinoma model [18]. Moreover, other studies have shown that clodronate liposomes enhance the anti-tumor and anti-angiogenic effects of anti-VEGF antibodies in subcutaneous tumor models [19][20].
2.2. Recruitment
The majority of monocyte recruitment inhibitors are monoclonal antibodies or pharmacological targets designed to block the interaction between monocyte-chemotactic cytokines and chemokines and their receptors. There are currently several clinical trials testing the safety and efficacy of such treatments as single agents or in combination with other therapies [6].
Much of the research investigating monocyte/macrophage recruitment strategies as targets for anti-cancer therapy have centered around the CCL2/CCR2 axis given that CCR2 is overexpressed by many tumors and enhances monocyte recruitment into the TME [21]. Indeed, use of anti-CCL2 antibodies have been associated with reduced macrophage infiltration and tumor growth [21][22]. However, in the clinic, completion of a Phase II clinical trial using anti-CCL2 antibodies in castration-resistant prostate cancer found that treatment neither blocked the signaling axis nor did it reduce tumor burden [23]. Therapeutic approaches targeting CCR2 by pharmacological inhibitors, such as PF-136309, anti-CCR2, or small-interfering RNA (siRNA) knockdown, are still under investigation [24][25][26]. As mentioned previously, many macrophage targeting therapies will be most efficacious when used in conjunction with others rather than when used alone. A recent study revealed that CCR2 antagonism sensitized tumors to anti-PD-1 ICB therapy in a number of murine tumor and metastasis models. In these models, tumor regression was associated with increased mobilization and activation of CD8+ T cells [27]. While single-agent strategies targeting monocyte recruitment may be beneficial in alleviating pro-tumorigenic effects of recruited macrophage populations, resident macrophage populations will remain undisturbed and may thwart attempts of regaining tumor control.
2.3. Repolarization
The abundance of macrophages, their contribution to the TME, and their plasticity make them attractive targets not only for depletion but also for repolarization strategies. As discussed below, many experimental therapies have focused on repolarizing these tumor-promoting cells towards a tumor-suppressive phenotype to improve patient outcomes.
Engagement of toll-like receptors (TLR) with their cognate ligands is a well-described method of inducing macrophage repolarization. TLR agonists, including lipopolysaccharides, and lipoproteins, stimulate NFκB, activator protein 1 (AP-1), and interferon regulatory factor signaling pathways. This results in activation of M1-associated genes and production of pro-inflammatory cytokines, including TNFα, IL-12, and IL-6 [28]. Indeed, Poly:IC-mediated activation of TLR-3 in a murine model of colon cancer led to M1 macrophage repolarization and reduced tumor growth through activation of IFN-α/β signaling pathways [29]. Despite successes in pre-clinical models, use of systemic TLR agonists as anti-cancer therapy is hindered by toxicity [30]. However, intratumoral TLR agonist therapy [31] or use of drug delivery systems to directly target TLRs expressed by TAMs can mitigate toxicity. For example, administration of β-cyclodextrin nanoparticles loaded with TLR7/8 agonists to tumor-bearing mice led to effective drug delivery to TAMs with limited off-target effects. These TLR7/8 agonist-loaded nanoparticles were potent inducers of M2-to-M1 macrophage repolarization and sensitized mice to anti-PD-1 therapy in PD-1 refractory tumors [32]. In addition, activation of the cytosolic stimulator of interferon genes (STING) pathway, which results in type-I interferon expression, has also been associated with macrophage polarization [33] and potentiation of ICB responses [34] in pre-clinical models.
3. Conclusions
There is accumulating evidence that macrophages are key players in cancer evolution and, consequently, patient outcomes. Therefore, macrophages are obvious candidates for therapeutic intervention strategies. However, a deeper understanding of the biological role of these cells is needed to design more effective treatment options for patients with cancer. Phenotypic and transcriptomic differences among macrophages of different origins provide a rationale to consider macrophage ontology when studying macrophage-mediated tumor promotional pathways and potential targeting options. Despite significant advancements in development and implementation of immune therapy approaches targeting macrophages, many obstacles still remain and continue to be a barrier to successful patient treatment. When should treatment begin? Which treatments or combinations thereof will achieve long-term tumor control? Will there be significant toxicities associated with combination therapy? How can we improve treatment delivery methods to induce a potent but tumor-localized therapy response? With advancing research, these outstanding questions, and many others, will hopefully be resolved.
This entry is adapted from the peer-reviewed paper 10.3390/cells10040960
References
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925.
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126.
- Pawelek, J.; Chakraborty, A.; Lazova, R.; Yilmaz, Y.; Cooper, D.; Brash, D.; Handerson, T. Co-opting macrophage traits in cancer progression: A consequence of tumor cell fusion? Contrib. Microbiol. 2006, 13, 138–155.
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382.
- Cassetta, L.; Kitamura, T. Targeting Tumor-Associated Macrophages as a Potential Strategy to Enhance the Response to Immune Checkpoint Inhibitors. Front. Cell Dev. Biol. 2018, 6, 38.
- Anfray, C.; Ummarino, A.; Andon, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2019, 9, 46.
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61.
- Strachan, D.C.; Ruffell, B.; Oei, Y.; Bissell, M.J.; Coussens, L.M.; Pryer, N.; Daniel, D. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8 T cells. Oncoimmunology 2013, 2, e26968.
- Fritz, J.M.; Tennis, M.A.; Orlicky, D.J.; Lin, H.; Ju, C.; Redente, E.F.; Choo, K.S.; Staab, T.A.; Bouchard, R.J.; Merrick, D.T.; et al. Depletion of tumor-associated macrophages slows the growth of chemically induced mouse lung adenocarcinomas. Front. Immunol. 2014, 5, 587.
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018.
- O’Brien, S.A.; Orf, J.; Skrzypczynska, K.M.; Tan, H.; Kim, J.; DeVoss, J.; Belmontes, B.; Egen, J.G. Activity of tumor-associated macrophage depletion by CSF1R blockade is highly dependent on the tumor model and timing of treatment. Cancer Immunol. Immunother. 2021.
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.M.; Ries, C.H.; Ruttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53.
- Tham, M.; Khoo, K.; Yeo, K.P.; Kato, M.; Prevost-Blondel, A.; Angeli, V.; Abastado, J.P. Macrophage depletion reduces postsurgical tumor recurrence and metastatic growth in a spontaneous murine model of melanoma. Oncotarget 2015, 6, 22857–22868.
- Rogers, T.L.; Holen, I. Tumour macrophages as potential targets of bisphosphonates. J. Transl. Med. 2011, 9, 177.
- Van Acker, H.H.; Anguille, S.; Willemen, Y.; Smits, E.L.; Van Tendeloo, V.F. Bisphosphonates for cancer treatment: Mechanisms of action and lessons from clinical trials. Pharmacol. Ther. 2016, 158, 24–40.
- Biewenga, J.; van der Ende, M.B.; Krist, L.F.; Borst, A.; Ghufron, M.; van Rooijen, N. Macrophage depletion in the rat after intraperitoneal administration of liposome-encapsulated clodronate: Depletion kinetics and accelerated repopulation of peritoneal and omental macrophages by administration of Freund’s adjuvant. Cell Tissue Res. 1995, 280, 189–196.
- Carron, E.C.; Homra, S.; Rosenberg, J.; Coffelt, S.B.; Kittrell, F.; Zhang, Y.; Creighton, C.J.; Fuqua, S.A.; Medina, D.; Machado, H.L. Macrophages promote the progression of premalignant mammary lesions to invasive cancer. Oncotarget 2017, 8, 50731–50746.
- Zhang, W.; Zhu, X.D.; Sun, H.C.; Xiong, Y.Q.; Zhuang, P.Y.; Xu, H.X.; Kong, L.Q.; Wang, L.; Wu, W.Z.; Tang, Z.Y. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin. Cancer Res. 2010, 16, 3420–3430.
- Priceman, S.J.; Sung, J.L.; Shaposhnik, Z.; Burton, J.B.; Torres-Collado, A.X.; Moughon, D.L.; Johnson, M.; Lusis, A.J.; Cohen, D.A.; Iruela-Arispe, M.L.; et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: Combating tumor evasion of antiangiogenic therapy. Blood 2010, 115, 1461–1471.
- Zeisberger, S.M.; Odermatt, B.; Marty, C.; Zehnder-Fjällman, A.H.M.; Ballmer-Hofer, K.; Schwendener, R.A. Clodronate-liposome-mediated depletion of tumour-associated macrophages: A new and highly effective antiangiogenic therapy approach. Br. J. Cancer 2006, 95, 272–281.
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 2016, 7, 28697–28710.
- Argyle, D.; Kitamura, T. Targeting Macrophage-Recruiting Chemokines as a Novel Therapeutic Strategy to Prevent the Progression of Solid Tumors. Front. Immunol. 2018, 9, 2629.
- Pienta, K.J.; Machiels, J.P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investig. New Drugs 2013, 31, 760–768.
- Sawa-Wejksza, K.; Kandefer-Szerszeń, M. Tumor-Associated Macrophages as Target for Antitumor Therapy. Arch. Immunol. Ther. Exp. 2018, 66, 97–111.
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662.
- Fang, W.B.; Yao, M.; Brummer, G.; Acevedo, D.; Alhakamy, N.; Berkland, C.; Cheng, N. Targeted gene silencing of CCL2 inhibits triple negative breast cancer progression by blocking cancer stem cell renewal and M2 macrophage recruitment. Oncotarget 2016, 7, 49349–49367.
- Tu, M.M.; Abdel-Hafiz, H.A.; Jones, R.T.; Jean, A.; Hoff, K.J.; Duex, J.E.; Chauca-Diaz, A.; Costello, J.C.; Dancik, G.M.; Tamburini, B.A.J.; et al. Inhibition of the CCL2 receptor, CCR2, enhances tumor response to immune checkpoint therapy. Commun. Biol. 2020, 3, 720.
- van Dalen, F.J.; van Stevendaal, M.; Fennemann, F.L.; Verdoes, M.; Ilina, O. Molecular Repolarisation of Tumour-Associated Macrophages. Molecules 2018, 24, 9.
- Vidyarthi, A.; Khan, N.; Agnihotri, T.; Negi, S.; Das, D.K.; Aqdas, M.; Chatterjee, D.; Colegio, O.R.; Tewari, M.K.; Agrewala, J.N. TLR-3 Stimulation Skews M2 Macrophages to M1 Through IFN-alphabeta Signaling and Restricts Tumor Progression. Front. Immunol. 2018, 9, 1650.
- Anwar, M.A.; Shah, M.; Kim, J.; Choi, S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med. Res. Rev. 2019, 39, 1053–1090.
- Mullins, S.R.; Vasilakos, J.P.; Deschler, K.; Grigsby, I.; Gillis, P.; John, J.; Elder, M.J.; Swales, J.; Timosenko, E.; Cooper, Z.; et al. Intratumoral immunotherapy with TLR7/8 agonist MEDI9197 modulates the tumor microenvironment leading to enhanced activity when combined with other immunotherapies. J. Immunother. Cancer 2019, 7, 244.
- Rodell, C.B.; Arlauckas, S.P.; Cuccarese, M.F.; Garris, C.S.; Li, R.; Ahmed, M.S.; Kohler, R.H.; Pittet, M.J.; Weissleder, R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018, 2, 578–588.
- Downey, C.M.; Aghaei, M.; Schwendener, R.A.; Jirik, F.R. DMXAA causes tumor site-specific vascular disruption in murine non-small cell lung cancer, and like the endogenous non-canonical cyclic dinucleotide STING agonist, 2’3’-cGAMP, induces M2 macrophage repolarization. PLoS ONE 2014, 9, e99988.
- Gadkaree, S.K.; Fu, J.; Sen, R.; Korrer, M.J.; Allen, C.; Kim, Y.J. Induction of tumor regression by intratumoral STING agonists combined with anti-programmed death-L1 blocking antibody in a preclinical squamous cell carcinoma model. Head Neck 2017, 39, 1086–1094.