Leishmania is a protozoan parasite of the trypanosomatid family, causing a wide range of diseases with different clinical manifestations including cutaneous, mucocutaneous and visceral leishmaniasis. According to WHO, one billion people are at risk of Leishmania infection as they live in endemic areas while there are 12 million infected people worldwide. Annually, 0.9–1.6 million new infections are reported and 20–50 thousand deaths occur due to Leishmania infection. As current chemotherapy for treating leishmaniasis exhibits numerous drawbacks and due to the lack of effective human vaccine, there is an urgent need to develop new antileishmanial therapy treatment. To this end, eukaryotic protein kinases can be ideal target candidates for rational drug design against leishmaniasis. Eukaryotic protein kinases mediate signal transduction through protein phosphorylation and their inhibition is anticipated to be disease modifying as they regulate all essential processes for Leishmania viability and completion of the parasitic life cycle including cell-cycle progression, differentiation and virulence. This review highlights existing knowledge concerning the exploitation of Leishmania protein kinases as molecular targets to treat leishmaniasis and the current knowledge of their role in the biology of Leishmania spp. and in the regulation of signalling events that promote parasite survival in the insect vector or the mammalian host.
- Leishmania
- leishmaniasis
- protein kinases
- cell cycle
- drug targets
- MAPKs
- CDKs
- GSK-3
- DYRKs
- trypanosomatids
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
Leishmaniases and Current Chemotherapy
2. Eukaryotic Protein Kinases (ePKs) as Potential Drug Targets
3. Protein Kinase Domains and Kinase Inhibition
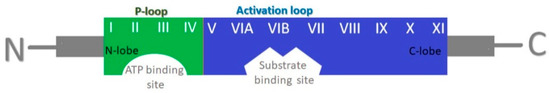
4. Cyclin-Dependent Kinases (CDKs)

5. Glycogen Synthase Kinase 3 (GSK-3)
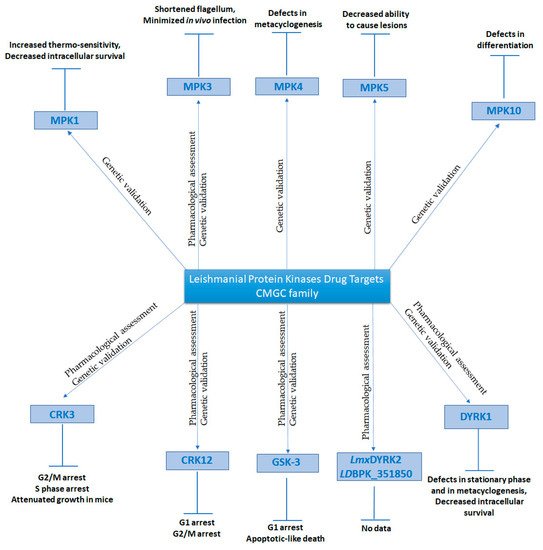
6. Dual-Specificity Tyrosine-Regulated Kinases (DYRKs)
7. Mitogen-activated Protein Kinases
8. Casein Kinases
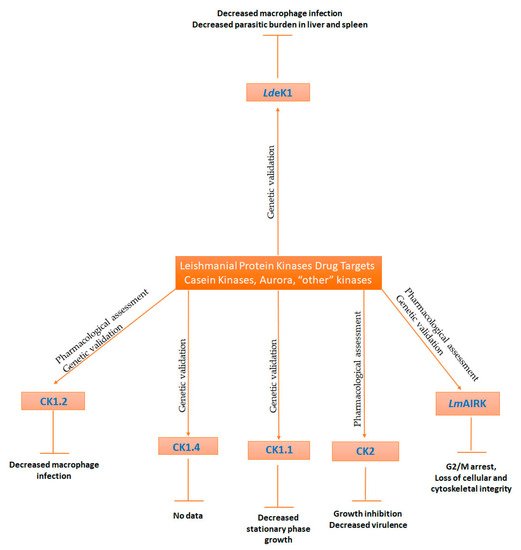
9. Aurora Kinases
10. Other Kinases
11. Future Directions—Concluding Remarks
Funding
Conflicts of Interest
References
- WHO. Leishmaniasis. Available online: https://www.who.int/health-topics/leishmaniasis#tab=tab_1 (accessed on 15 February 2021).
- Bates, P.A. The developmental biology of Leishmania promastigotes. Exp. Parasitol. 1994, 79, 215–218. [Google Scholar] [CrossRef]
- Sacks, D.L.; Perkins, P.V. Identification of an infective stage of Leishmania promastigotes. Science 1984, 223, 1417–1419. [Google Scholar] [CrossRef] [PubMed]
- Merritt, C.; Silva, L.E.; Tanner, A.L.; Stuart, K.; Pollastri, M.P. Kinases as druggable targets in trypanosomatid protozoan parasites. Chem. Rev. 2014, 114, 11280–11304. [Google Scholar] [CrossRef]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Colmenares, M.; Kar, S.; Goldsmith-Pestana, K.; McMahon-Pratt, D. Mechanisms of pathogenesis: Differences amongst Leishmania species. Trans. R Soc. Trop. Med. Hyg. 2002, 96 (Suppl. 1), S3–S7. [Google Scholar] [CrossRef]
- Zijlstra, E.E. PKDL and other dermal lesions in HIV co-infected patients with Leishmaniasis: Review of clinical presentation in relation to immune responses. PLoS Negl. Trop. Dis. 2014, 8, e3258. [Google Scholar] [CrossRef] [PubMed]
- Cincurá, C.; de Lima, C.M.F.; Machado, P.R.L.; Oliveira-Filho, J.; Glesby, M.J.; Lessa, M.M.; Carvalho, E.M. Mucosal leishmaniasis: A Retrospective Study of 327 Cases from an Endemic Area of Leishmania (Viannia) braziliensis. Am. J. Trop. Med. Hyg. 2017, 97, 761–766. [Google Scholar] [CrossRef]
- Maltezou, H.C. Drug resistance in visceral leishmaniasis. J. Biomed. Biotechnol. 2010, 2010, 617521. [Google Scholar] [CrossRef] [PubMed]
- Uliana, S.R.B.; Trinconi, C.T.; Coelho, A.C. Chemotherapy of leishmaniasis: Present challenges. Parasitology 2018, 145, 464–480. [Google Scholar] [CrossRef] [PubMed]
- Hailu, A.; Musa, A.; Wasunna, M.; Balasegaram, M.; Yifru, S.; Mengistu, G.; Hurissa, Z.; Hailu, W.; Weldegebreal, T.; Tesfaye, S.; et al. Geographical variation in the response of visceral leishmaniasis to paromomycin in East Africa: A multicentre, open-label, randomized trial. PLoS Negl. Trop. Dis. 2010, 4, e709. [Google Scholar] [CrossRef] [PubMed]
- Musa, A.; Khalil, E.; Hailu, A.; Olobo, J.; Balasegaram, M.; Omollo, R.; Edwards, T.; Rashid, J.; Mbui, J.; Musa, B.; et al. Sodium stibogluconate (SSG) & paromomycin combination compared to SSG for visceral leishmaniasis in East Africa: A randomised controlled trial. PLoS Negl. Trop. Dis. 2012, 6, e1674. [Google Scholar] [CrossRef]
- Jha, S.N.; Singh, N.K.; Jha, T.K. Changing response to diamidine compounds in cases of kala-azar unresponsive to antimonial. J. Assoc. Physicians India 1991, 39, 314–316. [Google Scholar]
- Rahman, M.; Ahmed, B.N.; Faiz, M.A.; Chowdhury, M.Z.; Islam, Q.T.; Sayeedur, R.; Rahman, M.R.; Hossain, M.; Bangali, A.M.; Ahmad, Z.; et al. Phase IV trial of miltefosine in adults and children for treatment of visceral leishmaniasis (kala-azar) in Bangladesh. Am. J. Trop. Med. Hyg. 2011, 85, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.K.; Sinha, P.K.; Sundar, S.; Thakur, C.P.; Jha, T.K.; Pandey, K.; Das, V.R.; Kumar, N.; Lal, C.; Verma, N.; et al. Phase 4 trial of miltefosine for the treatment of Indian visceral leishmaniasis. J. Infect. Dis. 2007, 196, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Dorlo, T.P.; van Thiel, P.P.; Huitema, A.D.; Keizer, R.J.; de Vries, H.J.; Beijnen, J.H.; de Vries, P.J. Pharmacokinetics of miltefosine in Old World cutaneous leishmaniasis patients. Antimicrob. Agents Chemother. 2008, 52, 2855–2860. [Google Scholar] [CrossRef]
- Bahrami, S.; Oryan, A.; Bemani, E. Efficacy of amiodarone and voriconazole combination therapy in cutaneous leishmaniasis in the mice experimentally infected with Leishmania major. J. Infect. Chemother. 2021. [Google Scholar] [CrossRef]
- Andrade-Neto, V.V.; Rebello, K.M.; Pereira, T.M.; Torres-Santos, E.C. Effect of Itraconazole-Ezetimibe-Miltefosine Ternary Therapy in Murine Visceral Leishmaniasis. Antimicrob. Agents Chemother. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Curtis, C.R.; Tur-Gracia, S.; Olatunji, T.O.; Carter, K.C.; Williams, R.A.M. Drug combinations as effective anti-leishmanials against drug resistant. RSC Med. Chem. 2020, 11, 905–912. [Google Scholar] [CrossRef]
- Gugliandolo, E.; Palma, E.; Peritore, A.F.; Siracusa, R.; D’Amico, R.; Fusco, R.; Licata, P.; Crupi, R. Effect of Artesunate on Leishmania Amazonesis Induced Neuroinflammation and Nociceptive Behavior in Male Balb/C Mice. Animals 2020, 10, 557. [Google Scholar] [CrossRef]
- Medkour, H.; Bitam, I.; Laidoudi, Y.; Lafri, I.; Lounas, A.; Hamidat, H.K.; Mekroud, A.; Varloud, M.; Davoust, B.; Mediannikov, O. Potential of Artesunate in the treatment of visceral leishmaniasis in dogs naturally infected by Leishmania infantum: Efficacy evidence from a randomized field trial. PLoS Negl. Trop. Dis. 2020, 14, e0008947. [Google Scholar] [CrossRef] [PubMed]
- Machín, L.; Nápoles, R.; Gille, L.; Monzote, L. Leishmania amazonensis response to artemisinin and derivatives. Parasitol. Int. 2021, 80, 102218. [Google Scholar] [CrossRef]
- Mutiso, J.M.; Macharia, J.C.; Barasa, M.; Taracha, E.; Bourdichon, A.J.; Gicheru, M.M. In vitro and in vivo antileishmanial efficacy of a combination therapy of diminazene and artesunate against Leishmania donovani in BALB/c mice. Rev. Inst. Med. Trop. Sao Paulo 2011, 53, 129–132. [Google Scholar] [CrossRef]
- Meijer, L.; Skaltsounis, A.L.; Magiatis, P.; Polychronopoulos, P.; Knockaert, M.; Leost, M.; Ryan, X.P.; Vonica, C.A.; Brivanlou, A.; Dajani, R.; et al. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem. Biol. 2003, 10, 1255–1266. [Google Scholar] [CrossRef]
- Grant, K.M.; Dunion, M.H.; Yardley, V.; Skaltsounis, A.L.; Marko, D.; Eisenbrand, G.; Croft, S.L.; Meijer, L.; Mottram, J.C. Inhibitors of Leishmania mexicana CRK3 cyclin-dependent kinase: Chemical library screen and antileishmanial activity. Antimicrob. Agents Chemother. 2004, 48, 3033–3042. [Google Scholar] [CrossRef] [PubMed]
- Xingi, E.; Smirlis, D.; Myrianthopoulos, V.; Magiatis, P.; Grant, K.M.; Meijer, L.; Mikros, E.; Skaltsounis, A.L.; Soteriadou, K. 6-Br-5methylindirubin-3′oxime (5-Me-6-BIO) targeting the leishmanial glycogen synthase kinase-3 (GSK-3) short form affects cell-cycle progression and induces apoptosis-like death: Exploitation of GSK-3 for treating leishmaniasis. Int. J. Parasitol. 2009, 39, 1289–1303. [Google Scholar] [CrossRef]
- Efstathiou, A.; Gaboriaud-Kolar, N.; Smirlis, D.; Myrianthopoulos, V.; Vougogiannopoulou, K.; Alexandratos, A.; Kritsanida, M.; Mikros, E.; Soteriadou, K.; Skaltsounis, A.L. An inhibitor-driven study for enhancing the selectivity of indirubin derivatives towards leishmanial Glycogen Synthase Kinase-3 over leishmanial cdc2-related protein kinase 3. Parasit. Vectors 2014, 7, 234. [Google Scholar] [CrossRef] [PubMed]
- Efstathiou, A.; Meira, C.S.; Gaboriaud-Kolar, N.; Bastos, T.M.; Rocha, V.P.C.; Vougogiannopoulou, K.; Skaltsounis, A.L.; Smirlis, D.; Soares, M.B.P. Indirubin derivatives are potent and selective anti-Trypanosoma cruzi agents. Virulence 2018, 9, 1658–1668. [Google Scholar] [CrossRef] [PubMed]
- Efstathiou, A.; Gaboriaud-Kolar, N.; Myrianthopoulos, V.; Vougogiannopoulou, K.; Subota, I.; Aicher, S.; Mikros, E.; Bastin, P.; Skaltsounis, A.L.; Soteriadou, K.; et al. Indirubin Analogues Inhibit Trypanosoma brucei Glycogen Synthase Kinase-3 Short and T. brucei Growth. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Karampetsou, K.; Koutsoni, O.S.; Gogou, G.; Angelis, A.; Skaltsounis, L.A.; Dotsika, E. Total Phenolic Fraction (TPF) from Extra Virgin Olive Oil: Induction of apoptotic-like cell death in Leishmania spp. promastigotes and in vivo potential of therapeutic immunomodulation. PLoS Negl. Trop. Dis. 2021, 15, e0008968. [Google Scholar] [CrossRef]
- Timmers, L.F.; Pauli, I.; Barcellos, G.B.; Rocha, K.B.; Caceres, R.A.; de Azevedo, W.F.; Soares, M.B. Genomic databases and the search of protein targets for protozoan parasites. Curr. Drug Targets 2009, 10, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Ivens, A.C.; Peacock, C.S.; Worthey, E.A.; Murphy, L.; Aggarwal, G.; Berriman, M.; Sisk, E.; Rajandream, M.A.; Adlem, E.; Aert, R.; et al. The genome of the kinetoplastid parasite, Leishmania major. Science 2005, 309, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Smirlis, D.; Soares, M.B. Selection of molecular targets for drug development against trypanosomatids. Subcell. Biochem. 2014, 74, 43–76. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.; Worthey, E.A.; Ward, P.N.; Mottram, J.C. Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, Trypanosoma brucei and Trypanosoma cruzi. BMC Genomics 2005, 6, 127. [Google Scholar] [CrossRef] [PubMed]
- Borba, J.V.B.; Silva, A.C.; Ramos, P.I.P.; Grazzia, N.; Miguel, D.C.; Muratov, E.N.; Furnham, N.; Andrade, C.H. Unveiling the Kinomes of Leishmania infantum and L. braziliensis Empowers the Discovery of New Kinase Targets and Antileishmanial Compounds. Comput. Struct. Biotechnol. J. 2019, 17, 352–361. [Google Scholar] [CrossRef]
- Baker, N.; Catta-Preta, C.M.C.; Neish, R.; Sadlova, J.; Powell, B.; Alves-Ferreira, E.V.C.; Geoghegan, V.; Carnielli, J.B.T.; Newling, K.; Hughes, C.; et al. Systematic functional analysis of Leishmania protein kinases identifies regulators of differentiation or survival. Nat. Commun. 2021, 12, 1244. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Hanks, S.K.; Hunter, T. Protein kinases 6. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 1995, 9, 576–596. [Google Scholar] [CrossRef]
- Knighton, D.R.; Zheng, J.H.; Ten Eyck, L.F.; Ashford, V.A.; Xuong, N.H.; Taylor, S.S.; Sowadski, J.M. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 1991, 253, 407–414. [Google Scholar] [CrossRef]
- Hubbard, S.R.; Wei, L.; Ellis, L.; Hendrickson, W.A. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 1994, 372, 746–754. [Google Scholar] [CrossRef]
- Brinkworth, R.I.; Breinl, R.A.; Kobe, B. Structural basis and prediction of substrate specificity in protein serine/threonine kinases. Proc. Natl. Acad. Sci. USA 2003, 100, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Nolen, B.; Taylor, S.; Ghosh, G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol. Cell 2004, 15, 661–675. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Knighton, D.R.; Zheng, J.; Ten Eyck, L.F.; Sowadski, J.M. Structural framework for the protein kinase family. Annu. Rev. Cell Biol. 1992, 8, 429–462. [Google Scholar] [CrossRef] [PubMed]
- Kobe, B.; Kemp, B.E. Principles of Kinase Regulation. In Handbook of Cell Signaling, 2/e; Elsevier Inc.: Cambridge, MA, USA, 2010; pp. 559–563. [Google Scholar]
- Böhmer, F.D.; Karagyozov, L.; Uecker, A.; Serve, H.; Botzki, A.; Mahboobi, S.; Dove, S. A single amino acid exchange inverts susceptibility of related receptor tyrosine kinases for the ATP site inhibitor STI-571. J. Biol. Chem. 2003, 278, 5148–5155. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.O. Cell cycle: Principles of control. Yale J. Biol. Med. 2007, 80, 141–142. [Google Scholar]
- Lohka, M.J.; Hayes, M.K.; Maller, J.L. Purification of maturation-promoting factor, an intracellular regulator of early mitotic events. Proc. Natl. Acad. Sci. USA 1988, 85, 3009–3013. [Google Scholar] [CrossRef]
- Nurse, P. Universal control mechanism regulating onset of M-phase. Nature 1990, 344, 503–508. [Google Scholar] [CrossRef]
- Dulić, V.; Lees, E.; Reed, S.I. Association of human cyclin E with a periodic G1-S phase protein kinase. Science 1992, 257, 1958–1961. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. G1 phase progression: Cycling on cue. Cell 1994, 79, 551–555. [Google Scholar] [CrossRef]
- Rosenblatt, J.; Gu, Y.; Morgan, D.O. Human cyclin-dependent kinase 2 is activated during the S and G2 phases of the cell cycle and associates with cyclin A. Proc. Natl. Acad. Sci. USA 1992, 89, 2824–2828. [Google Scholar] [CrossRef]
- Deshpande, A.; Sicinski, P.; Hinds, P.W. Cyclins and cdks in development and cancer: A perspective. Oncogene 2005, 24, 2909–2915. [Google Scholar] [CrossRef] [PubMed]
- Hassan, P.; Fergusson, D.; Grant, K.M.; Mottram, J.C. The CRK3 protein kinase is essential for cell cycle progression of Leishmania mexicana. Mol. Biochem. Parasitol. 2001, 113, 189–198. [Google Scholar] [CrossRef]
- Späth, G.F.; Clos, J. Joining forces: First application of a rapamycin-induced dimerizable Cre system for conditional null mutant analysis in Leishmania. Mol. Microbiol. 2016, 100, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.M.; Myburgh, E.; Philipon, C.; Brown, E.; Meissner, M.; Brewer, J.; Mottram, J.C. Conditional gene deletion with DiCre demonstrates an essential role for CRK3 in Leishmania mexicana cell cycle regulation. Mol. Microbiol. 2016, 100, 931–944. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.O.; Ibrahim, M.E.; Grant, K.M.; Mottram, J.C. Leishmania mexicana: Expression; characterization and activity assessment of E. coli-expressed recombinant CRK3. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 1338–1345. [Google Scholar] [PubMed]
- Gomes, F.C.; Ali, N.O.; Brown, E.; Walker, R.G.; Grant, K.M.; Mottram, J.C. Recombinant Leishmania mexicana CRK3:CYCA has protein kinase activity in the absence of phosphorylation on the T-loop residue Thr178. Mol. Biochem. Parasitol. 2010, 171, 89–96. [Google Scholar] [CrossRef]
- Banerjee, S.; Sen, A.; Das, P.; Saha, P. Leishmania donovani cyclin 1 (LdCyc1) forms a complex with cell cycle kinase subunit CRK3 (LdCRK3) and is possibly involved in S-phase-related activities. FEMS Microbiol. Lett. 2006, 256, 75–82. [Google Scholar] [CrossRef]
- Wang, Y.; Dimitrov, K.; Garrity, L.K.; Sazer, S.; Beverley, S.M. Stage-specific activity of the Leishmania major CRK3 kinase and functional rescue of a Schizosaccharomyces pombe cdc2 mutant. Mol. Biochem. Parasitol. 1998, 96, 139–150. [Google Scholar] [CrossRef]
- Řezníčková, E.; Popa, A.; Gucký, T.; Zatloukal, M.; Havlíček, L.; Bazgier, V.; Berka, K.; Jorda, R.; Popa, I.; Nasereddin, A.; et al. 2,6,9-Trisubstituted purines as CRK3 kinase inhibitors with antileishmanial activity in vitro. Bioorg. Med. Chem. Lett. 2015, 25, 2298–2301. [Google Scholar] [CrossRef]
- Cleghorn, L.A.; Woodland, A.; Collie, I.T.; Torrie, L.S.; Norcross, N.; Luksch, T.; Mpamhanga, C.; Walker, R.G.; Mottram, J.C.; Brenk, R.; et al. Identification of inhibitors of the Leishmania cdc2-related protein kinase CRK3. ChemMedChem 2011, 6, 2214–2224. [Google Scholar] [CrossRef]
- Walker, R.G.; Thomson, G.; Malone, K.; Nowicki, M.W.; Brown, E.; Blake, D.G.; Turner, N.J.; Walkinshaw, M.D.; Grant, K.M.; Mottram, J.C. High throughput screens yield small molecule inhibitors of Leishmania CRK3:CYC6 cyclin-dependent kinase. PLoS Negl. Trop. Dis. 2011, 5, e1033. [Google Scholar] [CrossRef] [PubMed]
- Maity, A.K.; Goswami, A.; Saha, P. Identification of substrates of an S-phase cell cycle kinase from Leishmania donovani. FEBS Lett. 2011, 585, 2635–2639. [Google Scholar] [CrossRef] [PubMed]
- Jorda, R.; Sacerdoti-Sierra, N.; Voller, J.; Havlíček, L.; Kráčalíková, K.; Nowicki, M.W.; Nasereddin, A.; Kryštof, V.; Strnad, M.; Walkinshaw, M.D.; et al. Anti-leishmanial activity of disubstituted purines and related pyrazolo[4,3-d]pyrimidines. Bioorg. Med. Chem. Lett. 2011, 21, 4233–4237. [Google Scholar] [CrossRef]
- Wyllie, S.; Thomas, M.; Patterson, S.; Crouch, S.; De Rycker, M.; Lowe, R.; Gresham, S.; Urbaniak, M.D.; Otto, T.D.; Stojanovski, L.; et al. Cyclin-dependent kinase 12 is a drug target for visceral leishmaniasis. Nature 2018, 560, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Mottram, J.C.; McCready, B.P.; Brown, K.G.; Grant, K.M. Gene disruptions indicate an essential function for the LmmCRK1 cdc2-related kinase of Leishmania mexicana. Mol. Microbiol. 1996, 22, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Grant, K.M.; Hassan, P.; Anderson, J.S.; Mottram, J.C. The crk3 gene of Leishmania mexicana encodes a stage-regulated cdc2-related histone H1 kinase that associates with p12. J. Biol. Chem. 1998, 273, 10153–10159. [Google Scholar] [CrossRef]
- Hoessel, R.; Leclerc, S.; Endicott, J.A.; Nobel, M.E.; Lawrie, A.; Tunnah, P.; Leost, M.; Damiens, E.; Marie, D.; Marko, D.; et al. Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat. Cell Biol. 1999, 1, 60–67. [Google Scholar] [CrossRef]
- Knockaert, M.; Wieking, K.; Schmitt, S.; Leost, M.; Grant, K.M.; Mottram, J.C.; Kunick, C.; Meijer, L. Intracellular Targets of Paullones. Identification following affinity purification on immobilized inhibitor. J. Biol. Chem. 2002, 277, 25493–25501. [Google Scholar] [CrossRef] [PubMed]
- Leost, M.; Schultz, C.; Link, A.; Wu, Y.Z.; Biernat, J.; Mandelkow, E.M.; Bibb, J.A.; Snyder, G.L.; Greengard, P.; Zaharevitz, D.W.; et al. Paullones are potent inhibitors of glycogen synthase kinase-3beta and cyclin-dependent kinase 5/p25. Eur. J. Biochem. 2000, 267, 5983–5994. [Google Scholar] [CrossRef]
- Martínez de Iturrate, P.; Sebastián-Pérez, V.; Nácher-Vázquez, M.; Tremper, C.S.; Smirlis, D.; Martín, J.; Martínez, A.; Campillo, N.E.; Rivas, L.; Gil, C. Towards discovery of new leishmanicidal scaffolds able to inhibit. J. Enzyme Inhib. Med. Chem. 2020, 35, 199–210. [Google Scholar] [CrossRef]
- Rachidi, N.; Taly, J.F.; Durieu, E.; Leclercq, O.; Aulner, N.; Prina, E.; Pescher, P.; Notredame, C.; Meijer, L.; Späth, G.F. Pharmacological assessment defines Leishmania donovani casein kinase 1 as a drug target and reveals important functions in parasite viability and intracellular infection. Antimicrob. Agents Chemother. 2014, 58, 1501–1515. [Google Scholar] [CrossRef]
- Durieu, E.; Prina, E.; Leclercq, O.; Oumata, N.; Gaboriaud-Kolar, N.; Vougogiannopoulou, K.; Aulner, N.; Defontaine, A.; No, J.H.; Ruchaud, S.; et al. From Drug Screening to Target Deconvolution: A Target-Based Drug Discovery Pipeline Using Leishmania Casein Kinase 1 Isoform 2 To Identify Compounds with Antileishmanial Activity. Antimicrob. Agents Chemother. 2016, 60, 2822–2833. [Google Scholar] [CrossRef]
- Zylbersztejn, A.M.; de Morais, C.G.; Lima, A.K.; Souza, J.E.; Lopes, A.H.; Da-Silva, S.A.; Silva-Neto, M.A.; Dutra, P.M. CK2 Secreted by Leishmania braziliensis Mediates Macrophage Association Invasion: A Comparative Study between Virulent and Avirulent Promastigotes. Biomed. Res. Int. 2015, 2015, 167323. [Google Scholar] [CrossRef]
- Dutra, P.M.; Vieira, D.P.; Meyer-Fernandes, J.R.; Silva-Neto, M.A.; Lopes, A.H. Stimulation of Leishmania tropica protein kinase CK2 activities by platelet-activating factor (PAF). Acta Trop. 2009, 111, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Loaëc, N.; Attanasio, E.; Villiers, B.; Durieu, E.; Tahtouh, T.; Cam, M.; Davis, R.A.; Alencar, A.; Roué, M.; Bourguet-Kondracki, M.L.; et al. Marine-Derived 2-Aminoimidazolone Alkaloids. Leucettamine B-Related Polyandrocarpamines Inhibit Mammalian and Protozoan DYRK & CLK Kinases. Mar. Drugs 2017, 15, 316. [Google Scholar] [CrossRef]
- Chhajer, R.; Bhattacharyya, A.; Didwania, N.; Shadab, M.; Das, N.; Palit, P.; Vaidya, T.; Ali, N. Leishmania donovani Aurora kinase: A promising therapeutic target against visceral leishmaniasis. Biochim. Biophys. Acta 2016, 1860, 1973–1988. [Google Scholar] [CrossRef]
- Patel, G.; Roncal, N.E.; Lee, P.J.; Leed, S.E.; Erath, J.; Rodriguez, A.; Sciotti, R.J.; Pollastri, M.P. Repurposing human Aurora kinase inhibitors as leads for anti-protozoan drug discovery. Medchemcomm 2014, 5, 655–658. [Google Scholar] [CrossRef] [PubMed]
- Raj, S.; Saha, G.; Sasidharan, S.; Dubey, V.K.; Saudagar, P. Biochemical characterization and chemical validation of Leishmania MAP Kinase-3 as a potential drug target. Sci. Rep. 2019, 9, 16209. [Google Scholar] [CrossRef] [PubMed]
- Horjales, S.; Schmidt-Arras, D.; Limardo, R.R.; Leclercq, O.; Obal, G.; Prina, E.; Turjanski, A.G.; Späth, G.F.; Buschiazzo, A. The crystal structure of the MAP kinase LmaMPK10 from Leishmania major reveals parasite-specific features and regulatory mechanisms. Structure 2012, 20, 1649–1660. [Google Scholar] [CrossRef]
- Nandan, D.; Zhang, N.; Yu, Y.; Schwartz, B.; Chen, S.; Kima, P.E.; Reiner, N.E. Miransertib (ARQ 092), an orally-available, selective Akt inhibitor is effective against Leishmania. PLoS ONE 2018, 13, e0206920. [Google Scholar] [CrossRef]
- Vacas, A.; Fernández-Rubio, C.; Algarabel, M.; Peña-Guerrero, J.; Larrea, E.; Rocha Formiga, F.; García-Sosa, A.T.; Nguewa, P.A. The Novel Serine/Threonine Protein Kinase LmjF.22.0810 from. Biomolecules 2019, 9, 723. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.P.; McNae, I.W.; Nowicki, M.W.; Zhong, W.; Michels, P.A.; Auld, D.S.; Fothergill-Gilmore, L.A.; Walkinshaw, M.D. The trypanocidal drug suramin and other trypan blue mimetics are inhibitors of pyruvate kinases and bind to the adenosine site. J. Biol Chem. 2011, 286, 31232–31240. [Google Scholar] [CrossRef]
- Morgan, H.P.; Walsh, M.J.; Blackburn, E.A.; Wear, M.A.; Boxer, M.B.; Shen, M.; Veith, H.; McNae, I.W.; Nowicki, M.W.; Michels, P.A.; et al. A new family of covalent inhibitors block nucleotide binding to the active site of pyruvate kinase. Biochem. J. 2012, 448, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Phukan, S.; Babu, V.S.; Kannoji, A.; Hariharan, R.; Balaji, V.N. GSK3beta: Role in therapeutic landscape and development of modulators. Br. J. Pharmacol. 2010, 160, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Ojo, K.K.; Gillespie, J.R.; Riechers, A.J.; Napuli, A.J.; Verlinde, C.L.; Buckner, F.S.; Gelb, M.H.; Domostoj, M.M.; Wells, S.J.; Scheer, A.; et al. Glycogen synthase kinase 3 is a potential drug target for African trypanosomiasis therapy. Antimicrob. Agents Chemother. 2008, 52, 3710–3717. [Google Scholar] [CrossRef]
- Ojo, K.K.; Arakaki, T.L.; Napuli, A.J.; Inampudi, K.K.; Keyloun, K.R.; Zhang, L.; Hol, W.G.; Verlinde, C.L.; Merritt, E.A.; Van Voorhis, W.C. Structure determination of glycogen synthase kinase-3 from Leishmania major and comparative inhibitor structure-activity relationships with Trypanosoma brucei GSK-3. Mol. Biochem. Parasitol. 2011, 176, 98–108. [Google Scholar] [CrossRef]
- Coghlan, M.P.; Culbert, A.A.; Cross, D.A.; Corcoran, S.L.; Yates, J.W.; Pearce, N.J.; Rausch, O.L.; Murphy, G.J.; Carter, P.S.; Roxbee Cox, L.; et al. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 2000, 7, 793–803. [Google Scholar] [CrossRef]
- Smith, D.G.; Buffet, M.; Fenwick, A.E.; Haigh, D.; Ife, R.J.; Saunders, M.; Slingsby, B.P.; Stacey, R.; Ward, R.W. 3-Anilino-4-arylmaleimides: Potent and selective inhibitors of glycogen synthase kinase-3 (GSK-3). Bioorg. Med. Chem. Lett. 2001, 11, 635–639. [Google Scholar] [CrossRef]
- Ryves, W.J.; Harwood, A.J. Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem. Biophys. Res. Commun. 2001, 280, 720–725. [Google Scholar] [CrossRef]
- Henriksen, E.J.; Kinnick, T.R.; Teachey, M.K.; O’Keefe, M.P.; Ring, D.; Johnson, K.W.; Harrison, S.D. Modulation of muscle insulin resistance by selective inhibition of GSK-3 in Zucker diabetic fatty rats. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E892–E900. [Google Scholar] [CrossRef]
- Kaidanovich-Beilin, O.; Eldar-Finkelman, H. Long-term treatment with novel glycogen synthase kinase-3 inhibitor improves glucose homeostasis in ob/ob mice: Molecular characterization in liver and muscle. J. Pharmacol. Exp. Ther. 2006, 316, 17–24. [Google Scholar] [CrossRef]
- Kannan, N.; Neuwald, A.F. Evolutionary constraints associated with functional specificity of the CMGC protein kinases MAPK, CDK, GSK, SRPK, DYRK, and CK2alpha. Protein. Sci. 2004, 13, 2059–2077. [Google Scholar] [CrossRef] [PubMed]
- Becker, W.; Joost, H.G. Structural and functional characteristics of Dyrk, a novel subfamily of protein kinases with dual specificity. Prog. Nucleic Acid. Res. Mol. Biol. 1999, 62, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Aranda, S.; Laguna, A.; de la Luna, S. DYRK family of protein kinases: Evolutionary relationships, biochemical properties, and functional roles. FASEB J. 2011, 25, 449–462. [Google Scholar] [CrossRef]
- Tejedor, F.; Zhu, X.R.; Kaltenbach, E.; Ackermann, A.; Baumann, A.; Canal, I.; Heisenberg, M.; Fischbach, K.F.; Pongs, O. minibrain: A new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron 1995, 14, 287–301. [Google Scholar] [CrossRef]
- Garrett, S.; Broach, J. Loss of Ras activity in Saccharomyces cerevisiae is suppressed by disruptions of a new kinase gene, YAKI, whose product may act downstream of the cAMP-dependent protein kinase. Genes Dev. 1989, 3, 1336–1348. [Google Scholar] [CrossRef]
- Garrett, S.; Menold, M.M.; Broach, J.R. The Saccharomyces cerevisiae YAK1 gene encodes a protein kinase that is induced by arrest early in the cell cycle. Mol. Cell. Biol. 1991, 11, 4045–4052. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.L.; Rodriguez-Gil, A.; Hornung, J. Integration of stress signals by homeodomain interacting protein kinases. Biol. Chem. 2014, 395, 375–386. [Google Scholar] [CrossRef]
- Huang, Y.; Deng, T.; Winston, B.W. Characterization of hPRP4 kinase activation: Potential role in signaling. Biochem. Biophys. Res. Commun. 2000, 271, 456–463. [Google Scholar] [CrossRef]
- Møller, R.S.; Kübart, S.; Hoeltzenbein, M.; Heye, B.; Vogel, I.; Hansen, C.P.; Menzel, C.; Ullmann, R.; Tommerup, N.; Ropers, H.H.; et al. Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am. J. Hum. Genet. 2008, 82, 1165–1170. [Google Scholar] [CrossRef]
- Park, J.; Song, W.J.; Chung, K.C. Function and regulation of Dyrk1A: Towards understanding Down syndrome. Cell Mol. Life Sci. 2009, 66, 3235–3240. [Google Scholar] [CrossRef]
- Kuhn, C.; Frank, D.; Will, R.; Jaschinski, C.; Frauen, R.; Katus, H.A.; Frey, N. DYRK1A is a novel negative regulator of cardiomyocyte hypertrophy. J. Biol. Chem. 2009, 284, 17320–17327. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ha, J.; Kim, H.J.; Kim, Y.S.; Chang, E.J.; Song, W.J.; Kim, H.H. Negative feedback Inhibition of NFATc1 by DYRK1A regulates bone homeostasis. J. Biol. Chem. 2009, 284, 33343–33351. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, S.R.; Jeong, H.K.; Radnaabazar, C.; Yoo, J.J.; Cho, H.J.; Lee, H.W.; Kim, I.S.; Cheon, Y.H.; Ahn, Y.S.; Chung, S.H.; et al. DYRK1A-mediated hyperphosphorylation of Tau. A functional link between Down syndrome and Alzheimer disease. J. Biol. Chem. 2007, 282, 34850–34857. [Google Scholar] [CrossRef] [PubMed]
- Friedman, E. Mirk/Dyrk1B in cancer. J. Cell Biochem. 2007, 102, 274–279. [Google Scholar] [CrossRef]
- Rocha, V.P.C.; Dacher, M.; Young, S.A.; Kolokousi, F.; Efstathiou, A.; Späth, G.F.; Soares, M.B.P.; Smirlis, D. Leishmania dual-specificity tyrosine-regulated kinase 1 (DYRK1) is required for sustaining Leishmania stationary phase phenotype. Mol. Microbiol. 2020, 113, 983–1002. [Google Scholar] [CrossRef]
- Murakami, N.; Bolton, D.; Hwang, Y.W. Dyrk1A binds to multiple endocytic proteins required for formation of clathrin-coated vesicles. Biochemistry 2009, 48, 9297–9305. [Google Scholar] [CrossRef]
- Murakami, N.; Bolton, D.C.; Kida, E.; Xie, W.; Hwang, Y.W. Phosphorylation by Dyrk1A of clathrin coated vesicle-associated proteins: Identification of the substrate proteins and the effects of phosphorylation. PLoS ONE 2012, 7, e34845. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Sun, X.; Goie, J.Y.G.; Zhang, Y. Regulation of Host Immune Responses against Influenza A Virus Infection by Mitogen-Activated Protein Kinases (MAPKs). Microorganisms 2020, 8, 1067. [Google Scholar] [CrossRef]
- Miyata, Y.; Akashi, M.; Nishida, E. Molecular cloning and characterization of a novel member of the MAP kinase superfamily. Genes Cells 1999, 4, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Ferrell, J.E.; Bhatt, R.R. Mechanistic studies of the dual phosphorylation of mitogen-activated protein kinase. J. Biol. Chem. 1997, 272, 19008–19016. [Google Scholar] [CrossRef]
- Naula, C.; Parsons, M.; Mottram, J.C. Protein kinases as drug targets in trypanosomes and Leishmania. Biochim. Biophys. Acta 2005, 1754, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Wiese, M. A mitogen-activated protein (MAP) kinase homologue of Leishmania mexicana is essential for parasite survival in the infected host. EMBO J. 1998, 17, 2619–2628. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Garg, M.; Hombach-Barrigah, A.; Clos, J.; Goyal, N. MAPK1 of Leishmania donovani interacts and phosphorylates HSP70 and HSP90 subunits of foldosome complex. Sci. Rep. 2017, 7, 10202. [Google Scholar] [CrossRef] [PubMed]
- Hombach-Barrigah, A.; Bartsch, K.; Smirlis, D.; Rosenqvist, H.; MacDonald, A.; Dingli, F.; Loew, D.; Späth, G.F.; Rachidi, N.; Wiese, M.; et al. Leishmania donovani 90 kD Heat Shock Protein—Impact of Phosphosites on Parasite Fitness, Infectivity and Casein Kinase Affinity. Sci. Rep. 2019, 9, 5074. [Google Scholar] [CrossRef]
- Morales, M.A.; Watanabe, R.; Dacher, M.; Chafey, P.; Osorio y Fortéa, J.; Scott, D.A.; Beverley, S.M.; Ommen, G.; Clos, J.; Hem, S.; et al. Phosphoproteome dynamics reveal heat-shock protein complexes specific to the Leishmania donovani infectious stage. Proc. Natl. Acad. Sci. USA 2010, 107, 8381–8386. [Google Scholar] [CrossRef]
- Garg, M.; Goyal, N. MAPK1 of Leishmania donovani modulates antimony susceptibility by downregulating P-glycoprotein efflux pumps. Antimicrob. Agents Chemother. 2015, 59, 3853–3863. [Google Scholar] [CrossRef]
- Mandal, G.; Sharma, M.; Kruse, M.; Sander-Juelch, C.; Munro, L.A.; Wang, Y.; Vilg, J.V.; Tamás, M.J.; Bhattacharjee, H.; Wiese, M.; et al. Modulation of Leishmania major aquaglyceroporin activity by a mitogen-activated protein kinase. Mol. Microbiol. 2012, 85, 1204–1218. [Google Scholar] [CrossRef]
- Goldman-Pinkovich, A.; Balno, C.; Strasser, R.; Zeituni-Molad, M.; Bendelak, K.; Rentsch, D.; Ephros, M.; Wiese, M.; Jardim, A.; Myler, P.J.; et al. An Arginine Deprivation Response Pathway Is Induced in Leishmania during Macrophage Invasion. PLoS Pathog. 2016, 12, e1005494. [Google Scholar] [CrossRef]
- Erdmann, M.; Scholz, A.; Melzer, I.M.; Schmetz, C.; Wiese, M. Interacting protein kinases involved in the regulation of flagellar length. Mol. Biol. Cell 2006, 17, 2035–2045. [Google Scholar] [CrossRef]
- Bengs, F.; Scholz, A.; Kuhn, D.; Wiese, M. LmxMPK9, a mitogen-activated protein kinase homologue affects flagellar length in Leishmania mexicana. Mol. Microbiol. 2005, 55, 1606–1615. [Google Scholar] [CrossRef]
- Wiese, M.; Kuhn, D.; Grünfelder, C.G. Protein kinase involved in flagellar-length control. Eukaryot. Cell 2003, 2, 769–777. [Google Scholar] [CrossRef]
- Wernimont, A.K.; Walker, J.R.; Hutchinson, A.; Loppnau, P.; Edwards, A.M.; Arrowsmith, C.H.; Bountra, C.; Hui, R.; Mangos, M. Crystal Structure of MPK3 from Leishmania Donovani, LdBPK_100540 in the Presence of NVP-BBT594. Available online: https://www.rcsb.org/structure/4O2Z (accessed on 15 February 2021).
- Herrera-Acevedo, C.; Dos Santos Maia, M.; Cavalcanti, É.; Coy-Barrera, E.; Scotti, L.; Scotti, M.T. Selection of antileishmanial sesquiterpene lactones from SistematX database using a combined ligand-/structure-based virtual screening approach. Mol. Divers. 2020. [Google Scholar] [CrossRef]
- Morales, M.A.; Renaud, O.; Faigle, W.; Shorte, S.L.; Späth, G.F. Over-expression of Leishmania major MAP kinases reveals stage-specific induction of phosphotransferase activity. Int. J. Parasitol. 2007, 37, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.A.; Pescher, P.; Späth, G.F. Leishmania major MPK7 protein kinase activity inhibits intracellular growth of the pathogenic amastigote stage. Eukaryot. Cell 2010, 9, 22–30. [Google Scholar] [CrossRef]
- Wang, Q.; Melzer, I.M.; Kruse, M.; Sander-Juelch, C.; Wiese, M. LmxMPK4, a mitogen-activated protein (MAP) kinase homologue essential for promastigotes and amastigotes of Leishmania mexicana. Kinetoplastid Biol. Dis. 2005, 4, 6. [Google Scholar] [CrossRef]
- Dacher, M.; Morales, M.A.; Pescher, P.; Leclercq, O.; Rachidi, N.; Prina, E.; Cayla, M.; Descoteaux, A.; Späth, G.F. Probing druggability and biological function of essential proteins in Leishmania combining facilitated null mutant and plasmid shuffle analyses. Mol. Microbiol. 2014, 93, 146–166. [Google Scholar] [CrossRef] [PubMed]
- John von Freyend, S.; Rosenqvist, H.; Fink, A.; Melzer, I.M.; Clos, J.; Jensen, O.N.; Wiese, M. LmxMPK4, an essential mitogen-activated protein kinase of Leishmania mexicana is phosphorylated and activated by the STE7-like protein kinase LmxMKK5. Int. J. Parasitol. 2010, 40, 969–978. [Google Scholar] [CrossRef]
- Raj, S.; Sasidharan, S.; Dubey, V.K.; Saudagar, P. Identification of lead molecules against potential drug target protein MAPK4 from L. donovani: An in-silico approach using docking, molecular dynamics and binding free energy calculation. PLoS ONE 2019, 14, e0221331. [Google Scholar] [CrossRef]
- Saravanan, P.; Venkatesan, S.K.; Mohan, C.G.; Patra, S.; Dubey, V.K. Mitogen-activated protein kinase 4 of Leishmania parasite as a therapeutic target. Eur. J. Med. Chem. 2010, 45, 5662–5670. [Google Scholar] [CrossRef] [PubMed]
- Wanders, P. In Vitro and In Vivo Characterization of a L. mexicana Lmx MPK5 Null Mutant. Master’s Thesis, Technische Hochschule Hannover, Hannover, Germany, 2004. [Google Scholar]
- Cayla, M.; Rachidi, N.; Leclercq, O.; Schmidt-Arras, D.; Rosenqvist, H.; Wiese, M.; Späth, G.F. Transgenic analysis of the Leishmania MAP kinase MPK10 reveals an auto-inhibitory mechanism crucial for stage-regulated activity and parasite viability. PLoS Pathog. 2014, 10, e1004347. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Pinna, L.A. Casein kinases as potential therapeutic targets. Expert Opin. Ther. Targets 2016, 20, 319–340. [Google Scholar] [CrossRef]
- Knippschild, U.; Gocht, A.; Wolff, S.; Huber, N.; Löhler, J.; Stöter, M. The casein kinase 1 family: Participation in multiple cellular processes in eukaryotes. Cell Signal. 2005, 17, 675–689. [Google Scholar] [CrossRef]
- Eide, E.J.; Virshup, D.M. Casein kinase I: Another cog in the circadian clockworks. Chronobiol. Int. 2001, 18, 389–398. [Google Scholar] [CrossRef]
- Price, M.A. CKI, there’s more than one: Casein kinase I family members in Wnt and Hedgehog signaling. Genes Dev. 2006, 20, 399–410. [Google Scholar] [CrossRef]
- Aoki, K.; Yoshida, K. Biological Consequences of Priming Phosphorylation in Cancer Development. Protein Phosphorylation 2017. [Google Scholar] [CrossRef]
- Flotow, H.; Graves, P.R.; Wang, A.Q.; Fiol, C.J.; Roeske, R.W.; Roach, P.J. Phosphate groups as substrate determinants for casein kinase I action. J. Biol. Chem. 1990, 265, 14264–14269. [Google Scholar] [CrossRef]
- Götz, C.; Montenarh, M. Protein kinase CK2 in development and differentiation. Biomed. Rep. 2017, 6, 127–133. [Google Scholar] [CrossRef]
- Sacerdoti-Sierra, N.; Jaffe, C.L. Release of ecto-protein kinases by the protozoan parasite Leishmania major. J. Biol. Chem. 1997, 272, 30760–30765. [Google Scholar] [CrossRef]
- Silverman, J.M.; Chan, S.K.; Robinson, D.P.; Dwyer, D.M.; Nandan, D.; Foster, L.J.; Reiner, N.E. Proteomic analysis of the secretome of Leishmania donovani. Genome Biol. 2008, 9, R35. [Google Scholar] [CrossRef]
- Silverman, J.M.; Clos, J.; de’Oliveira, C.C.; Shirvani, O.; Fang, Y.; Wang, C.; Foster, L.J.; Reiner, N.E. An exosome-based secretion pathway is responsible for protein export from Leishmania and communication with macrophages. J. Cell Sci. 2010, 123, 842–852. [Google Scholar] [CrossRef]
- Liu, J.; Carvalho, L.P.; Bhattacharya, S.; Carbone, C.J.; Kumar, K.G.; Leu, N.A.; Yau, P.M.; Donald, R.G.; Weiss, M.J.; Baker, D.P.; et al. Mammalian casein kinase 1alpha and its leishmanial ortholog regulate stability of IFNAR1 and type I interferon signaling. Mol. Cell. Biol. 2009, 29, 6401–6412. [Google Scholar] [CrossRef]
- Allocco, J.J.; Donald, R.; Zhong, T.; Lee, A.; Tang, Y.S.; Hendrickson, R.C.; Liberator, P.; Nare, B. Inhibitors of casein kinase 1 block the growth of Leishmania major promastigotes in vitro. Int. J. Parasitol. 2006, 36, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Dan-Goor, M.; Nasereddin, A.; Jaber, H.; Jaffe, C.L. Identification of a secreted casein kinase 1 in Leishmania donovani: Effect of protein over expression on parasite growth and virulence. PLoS ONE 2013, 8, e79287. [Google Scholar] [CrossRef]
- Martel, D.; Beneke, T.; Gluenz, E.; Späth, G.F.; Rachidi, N. Characterisation of Casein Kinase 1.1 in Leishmania donovani Using the CRISPR Cas9 Toolkit. Biomed. Res. Int 2017, 2017, 4635605. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Jaffe, C.L. Effect of protein kinase inhibitors on the growth, morphology, and infectivity of Leishmania promastigotes. Parasitol. Res. 1997, 83, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Vieira, L.L.; Sacerdoti-Sierra, N.; Jaffe, C.L. Effect of pH and temperature on protein kinase release by Leishmania donovani. Int J. Parasitol. 2002, 32, 1085–1093. [Google Scholar] [CrossRef]
- Silva-Neto, M.A.; Carneiro, A.B.; Vieira, D.P.; Mesquita, R.D.; Lopes, A.H. Platelet-activating factor (PAF) activates casein kinase 2 in the protozoan parasite Herpetomonas muscarum muscarum. Biochem. Biophys. Res. Commun. 2002, 293, 1358–1363. [Google Scholar] [CrossRef]
- Crane, R.; Gadea, B.; Littlepage, L.; Wu, H.; Ruderman, J.V. Aurora A, meiosis and mitosis. Biol. Cell 2004, 96, 215–229. [Google Scholar] [CrossRef]
- Tu, X.; Kumar, P.; Li, Z.; Wang, C.C. An aurora kinase homologue is involved in regulating both mitosis and cytokinesis in Trypanosoma brucei. J. Biol. Chem. 2006, 281, 9677–9687. [Google Scholar] [CrossRef] [PubMed]
- Siman-Tov, M.M.; Ivens, A.C.; Jaffe, C.L. Identification and cloning of Lmairk, a member of the Aurora/Ipl1p protein kinase family, from the human protozoan parasite Leishmania. Biochim. Biophys. Acta 2001, 1519, 241–245. [Google Scholar] [CrossRef]
- Reininger, L.; Wilkes, J.M.; Bourgade, H.; Miranda-Saavedra, D.; Doerig, C. An essential Aurora-related kinase transiently associates with spindle pole bodies during Plasmodium falciparum erythrocytic schizogony. Mol. Microbiol. 2011, 79, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Varela-M, R.E.; Ochoa, R.; Muskus, C.E.; Muro, A.; Mollinedo, F. Identification of a RAC/AKT-like gene in Leishmania parasites as a putative therapeutic target in leishmaniasis. Parasit. Vectors 2017, 10, 458. [Google Scholar] [CrossRef]
- Tirado-Duarte, D.; Marín-Villa, M.; Ochoa, R.; Blandón-Fuentes, G.; Soares, M.J.; Robledo, S.M.; Varela-Miranda, R.E. The Akt-like kinase of Leishmania panamensis: As a new molecular target for drug discovery. Acta Trop. 2018, 177, 171–178. [Google Scholar] [CrossRef]
- Jha, M.K.; Rao, S.J.; Sarode, A.Y.; Saha, B.; Kar, A.; Pal, J.K. A Leishmania donovani dominant-negative mutant for eIF2α kinase LdeK1 elicits host-protective immune response. Parasite Immunol. 2020, 42, e12678. [Google Scholar] [CrossRef]
- Abhishek, K.; Sardar, A.H.; Das, S.; Kumar, A.; Ghosh, A.K.; Singh, R.; Saini, S.; Mandal, A.; Verma, S.; Purkait, B.; et al. Phosphorylation of Translation Initiation Factor 2-Alpha in Leishmania donovani under Stress Is Necessary for Parasite Survival. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef]
- Srivastava, A.; Garg, S.; Jain, R.; Ayana, R.; Kaushik, H.; Garg, L.; Pati, S.; Singh, S. Identification and functional characterization of a bacterial homologue of Zeta toxin in Leishmania donovani. FEBS Lett. 2019, 593, 1223–1235. [Google Scholar] [CrossRef]
This entry is adapted from the peer-reviewed paper 10.3390/microorganisms9040691