Intracellular Ca2+ signaling is a major signal transduction pathway in non-excitable cells, responsible for the regulation of a variety of physiological functions. In the secretory epithelial cells of the exocrine pancreas, such as acinar and ductal cells, intracellular Ca2+ elevation regulates digestive enzyme secretion in acini or fluid and ion secretion in ductal cells. Although Ca2+ is a uniquely versatile orchestrator of epithelial physiology, unregulated global elevation of the intracellular Ca2+ concentration is an early trigger for the development of acute pancreatitis. Regardless of the aetiology, different forms of acute pancreatitis all exhibit sustained intracellular Ca2+ elevation as a common hallmark.
1. Ca2+ Signalling and Acute Pancreatitis
Acute pancreatitis (AP) is an inflammatory disease of the pancreas associated with significant morbidity and mortality. AP is one of the most frequent causes of hospitalisation among non-malignant gastrointestinal disorders, whereas the global disease incidence has increased in the past several decades [
1,
2,
3]. Although overall disease mortality has decreased in recent years [
4], the mortality from severe forms of AP (comprising about 10% of all cases) remains remarkably high (~28%) [
5]. AP is primarily caused by impacted gallstones or heavy alcohol consumption; however, the incidence of iatrogenic AP caused by endoscopic retrograde cholangiopancreatography (ERCP) or drug administration (such as L-asparaginase) has also increased [
5]. Regardless of the different pathogenic factors that may lead to acute inflammation of the pancreas, all cases are associated with sustained elevated intracellular Ca
2+ levels, which are a hallmark of AP pathogenesis [
6]. Intracellular Ca
2+ overload can lead to premature trypsinogen activation [
7,
8], mitochondrial damage and cell necrosis in pancreatic acinar cells [
9]. Our group has reported that sustained elevation of intracellular Ca
2+ in ductal cells impairs fluid and HCO
3- secretion [
10,
11], which are key functions of ductal cells, and also triggers mitochondrial damage with consequent ATP depletion and cell damage [
12]. As detailed below, toxins induce Ca
2+ release from the endoplasmic reticulum (ER) intracellular Ca
2+ stores, which is considered as the initial step in the development of sustained Ca
2+ elevation. Inhibition of the uncaged IP
3-induced Ca
2+ release by caffeine and other non-xanthine phosphodiesterase inhibitors leads to the impairment of toxin-induced Ca
2+ release and prevents mitochondrial depolarisation and acinar cell necrosis. It also improves the severity of cerulein, bile acid or fatty acid ethyl ester (FAEE)-induced experimental AP in mice [
13]. Sustained Ca
2+ signals can lead to mitochondrial injury, which can activate both apoptosis and necrosis. In the pathogenesis of AP opening of the mitochondrial permeability transition pore (MPTP) triggered by sustained elevated Ca
2+ levels is an initiating step in mitochondrial membrane potential (ΔΨ
m) loss, impaired mitochondrial ATP synthesis and increased permeability of the inner mitochondrial membrane, resulting in mitochondrial swelling and necrosis [
14,
15]. Genetic or pharmacologic inhibition of the MPTP resulted in the improvement of the AP phenotype and acinar [
16] and ductal [
17] cell injury both in vitro and in vivo.
1.1. Intracellular Ca2+ in Biliary AP
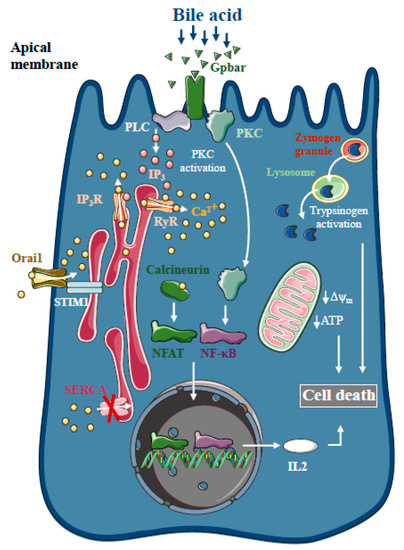
Bile acids are known to trigger dose-dependent elevations in intracellular Ca
2+ concentrations in isolated pancreatic acinar [
18] and ductal cells [
19] in vitro, which is due to Ca
2+ release from intracellular stores via the activation of IP
3 and ryanodine receptors, sarco-endoplasmic reticulum Ca
2+ pump (SERCA) inhibition [
20] and extracellular Ca
2+ influx activation [
21] (). The toxic effects of bile acids on pancreatic acinar cells also involve the activation of the G-protein-coupled cell surface bile acid receptor (Gpbar1) at the apical membrane, which also contributes to the development of sustained Ca
2+ elevation and its downstream effects [
22]. On the other hand, Gpbar1 knockout mice were protected against taurolithocholic acid 3-sulfate-induced AP, an experimental model of biliary AP.
Figure 1. Intracellular Ca2+ signalling in biliary acute pancreatitis. Bile acids dose-dependently release Ca2+ from intracellular stores via activation of IP3 and ryanodine receptors (RyR). The inhibition of the sarco-endoplasmic reticulum Ca2+ pump (SERCA) and activation of Orai1-mediated extracellular Ca2+ influx contributes to the sustained global Ca2+ signals. Bile acids can activate the G-protein-coupled cell surface bile acid receptor (Gpbar) at the apical membrane of pancreatic acinar cells that also release Ca2+ from the endoplasmic reticulum. Mitochondrial Ca2+ overload can lead to mitochondrial damage by opening the mitochondrial permeability transition pore and dissipating the mitochondrial membrane potential. In addition, bile acids have been demonstrated to activate calcineurin in a Ca2+-dependent manner in pancreatic cells, leading to premature digestive enzyme and NF-κB activation.
The pancreatic ductal epithelia can be another possible target of bile acids in the exocrine pancreas. Previous reports have suggested that increases in intracellular Ca
2+ in pancreatic ductal epithelial cells leads to a marked dose-dependent decrease in HCO
3- secretion upon bile acid exposure of isolated pancreatic ductal fragments [
19]. In addition, bile acids inhibit intracellular ATP production and decreased ΔΨ
m in acinar [
23,
24] and ductal cells [
12]. Interestingly, bile acid toxicity was not abolished by the removal of intracellular Ca
2+ elevation using a Ca
2+ chelator, BAPTA-AM, neither in acinar [
23] nor in ductal cells [
19], suggesting the existence of other parallel Ca
2+-independent effects of bile acids on the mitochondria. Besides inducing mitochondrial toxicity, bile acids have also been shown to activate calcineurin in a Ca
2+-dependent manner in pancreatic cells, leading to premature digestive enzyme and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation [
25]. The inhibition of calcineurin pharmacologically or by genetic knockout, reduced the severity of taurolithocholate 3-sulfate (TLCS)-induced AP and impaired protein kinase C, an upstream regulator of NF-κB activation and translocation [
26].
1.2. Intracellular Ca2+ in Alcoholic AP
Heavy alcohol consumption is the second most frequent cause of AP [
1]. As only a minority of alcoholics develop AP, genetic factors seem to play a major role in disease pathogenesis [
27]. However, direct treatment with ethanol and different non-oxidative ethanol metabolites have a damaging effect on acinar and ductal cells. In contrast to other organs, such as the liver, non-oxidative ethanol metabolism is the dominant metabolic pathway in the pancreas [
28], which is mediated by enzymes with FAEE synthase activity [
29], which combine ethanol and fatty acids to generate FAEE [
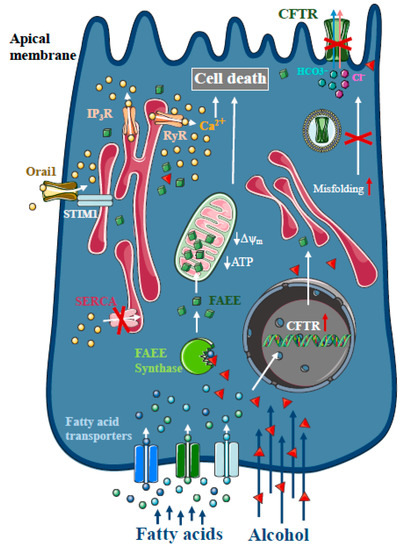
30]. Early studies suggested that more than 70% of FAEEs are preferentially accumulated in the mitochondria in cardiomyocytes, which is the site of fatty acid hydrolysis [
31]. Huang et al. investigated FAEE hydrolysis in pancreatic acinar cells using a fluorescently tagged palmitoleic acid probe that releases fluorescein upon hydrolysis [
32]. Their results suggest that FAEEs accumulate in mitochondria and that their local breakdown leads to high concentrations of fatty acids. Like bile acids, FAEEs have been shown to induce sustained [Ca
2+]
i elevation and reduced levels of cellular ATP, leading to necrosis [
9,
33,
34] (). Our group has demonstrated previously that alcohol and fatty acids inhibit fluid and HCO
3- secretion in the pancreatic ductal epithelia, mainly due to impaired expression and function of the cystic fibrosis transmembrane conductance regulator (CFTR) [
35], which was restored by ATP supplementation [
36]. Inhibition of CFTR activity was mediated by sustained intracellular Ca
2+ elevation, decreased adenosine 3′,5′-cyclic monophosphate (AMP) levels and impaired ATP production accompanied by ΔΨ
m depolarisation. Ethanol has reduced CFTR expression via accelerated plasma membrane turnover and impaired CFTR membrane stability. These alterations in the ductal cells increased the severity of alcohol-induced AP in mice.
Figure 2. Intracellular Ca2+ signalling in alcoholic acute pancreatitis. In the pancreas, non-oxidative ethanol metabolism is the dominant metabolic pathway mediated by enzymes with fatty acid ethyl ester (FAEE) synthase activity, which combine ethanol and fatty acids to generate FAEE. In pancreatic acinar cells, FAEEs are accumulated in the mitochondria and their local breakdown leads to localised high concentrations of fatty acids. Similar to bile acids, FAEEs induce sustained [Ca2+]i elevation and drop of cellular ATP leading to necrosis. In addition, alcohol and fatty acids inhibit fluid and HCO3- secretion in the pancreatic ductal epithelia, mainly due to the impaired expression and function of the cystic fibrosis transmembrane conductance regulator (CFTR).
1.3. Intracellular Ca2+ in Drug-Induced AP
AP is a relatively frequent complication of medical treatments, leading to painful inflammation and hospitalisation, and may serve as an indication to alter or cease otherwise effective therapy. Asparaginase, a long-term medication used to treat acute lymphoblastic leukaemia (ALL) in children, is one of the most likely drugs to induce AP. The incidence of asparaginase-induced AP is above 10% [
37], making it one of the most common causes for halting therapy for ALL [
38]. Recently, Peng et al. demonstrated that the treatment of isolated pancreatic acinar cell clusters with asparaginase triggers similar changes in intracellular Ca
2+ signalling as bile acids and ethanol metabolites [
39]. They showed that asparaginase treatment leads to intracellular Ca
2+ release, followed by extracellular Ca
2+ entry activation. Ca
2+ extrusion was also severely impaired, due to decreased intracellular ATP production, leading to cell necrosis. The inhibition of protease-activated receptor 2 (PAR2) abolished the toxic intracellular Ca
2+ signals, suggesting that the toxic effect of asparaginase is mediated by PAR2 activation. In addition, inhibition of the Ca
2+ entry by the selective Orai1 inhibitor, GSK-7975A, protected acinar cells from Ca
2+-induced damage. Similar effects were observed in vitro and in vivo by replacing glucose with galactose, which prevented the loss of ATP and protected acinar cells from necrosis [
40].
This entry is adapted from the peer-reviewed paper 10.3390/ijms21114005