Complement 5 (C5) protein, the cleavage of which is mediated by the serine proteases C5 convertases to generate two different fragments: C5a, which is a potent chemoattractant and pro-inflammatory modulator, and C5b, which initiates the formation of MAC, mediating cell lysis and triggering inflammation.
- complement system
- C5a/C5aR axis
- C5a receptor1
- neuropathies
- C5a inhibitor
1. Introduction
The complement system is a crucial element of the innate immune response that works in concert with antibodies and phagocytic cells to clear pathogens [1]. It consists of a number of precursor proteins that are cleaved by specific proteases to generate various complement peptides and fragments, ultimately leading to the formation of the Membrane Attack Complex (MAC) [2]. One of the key components of the complement system is the Complement 5 (C5) protein, the cleavage of which is mediated by the serine proteases C5 convertases to generate two different fragments [3]: C5a, which is a potent chemoattractant and pro-inflammatory modulator [4][5], and C5b, which initiates the formation of MAC, mediating cell lysis and triggering inflammation [6]. When properly activated, C5a is crucial for host defence system and clearance of pathogens; however, its inappropriate activation is involved in a wide range of disorders, including peripheral neuropathic diseases [7][8].
2. Complement Pathways
The complement is a major component of the innate immune system and acts as a bridge between innate and acquired immunity. Over the years, it has become clear that the complement has various functions, ranging from the mediation of inflammatory responses to the regulation of host cell clearance after their programmed cell death [9], and takes part in nearly every step of the immune reaction. It is composed of over 50 proteins [10]. Among these, the soluble ones are produced mainly by the liver and can be detected in the plasma and on cell surfaces as inactive precursors (zymogens) [1]; their cleavage by serine proteases activates a cascade of enzymatic reactions that is tightly regulated to assure complement activation is triggered only at specific locations, thus avoiding host tissue damage.
Activation of the complement system occurs through three distinct pathways: the classical (CP), lectin (LP), and alternative (AP) pathways [2] (Figure 1).
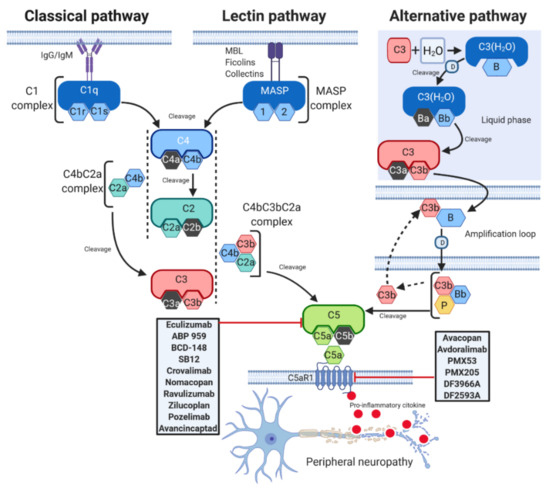
Figure 1. C5a complement activation pathways. The C5a complement system can be activated through three pathways: classical (CP), lectin (LP), and alternative (AP). CP begins with antibody-mediated activation of C1 complex, which leads to formation of the C4bC2a complex, the C3 convertase. This C3 convertase cleaves C3 to produce C3b, which forms a complex with C4b and C2a. This complex is the C5 convertase, which cleaves C5 to produce C5a and C5b. The LP begins with signal recognition by oligomeric structures of mannose- binding lectin (MBL), ficolins and collectins, which activate mannan-binding lectin serine proteases (MASP) 1 and 2, which in turn mediate the production of C4b. From this point, the LP follows the same steps as the CP. In the AP, C3 interacts with factor B (B) and factor D (D), leading to cleavage of further C3, and this process is perpetuated through an amplification loop. In the final step of this pathway, even properdin (P) is involved. Additional C3b binds to the C3 convertase and forms a C5 convertase, which cleaves C5 to form C5a and C5b. C5a activates on C5aR1, a prototypical G-protein coupled receptor (GPCR) recruiting immune cells to the site of inflammation. Drugs targeting C5 or C5aR1 in different stage of development are reported in the gray rectangles. T-arrow indicates inhibition of pathway at point of intersection.
Although each of them is differentially initiated and is characterised by unique and specific factors, they all include the activation of C3 and C5 and lead to a common pathway, which results in the formation of the MAC, ultimately inducing cell lysis by binding to the target cell membrane [11]. The CP is activated by the binding between Immunoglobulins M or G (IgM or IgG), and several other proteins such as C-reactive protein and serum amyloid P protein [12], and C1 complex, which is constituted by the sensing molecule C1q and two heterodimers formed by the zymogens C1r and C1s [12]. C1q activates C1r, which in turn cleaves C1s [13]. Once activated, C1 enzyme complex mediates the cleavage of native C4, which is followed by cleavage of C2 and the subsequent formation of the C3 convertase C4bC2a. The C3 convertase activates C3, triggering the dissociation of C3 into C3a and C3b. C3b then binds to the existing C3 convertase to form the C5 convertase C4bC3bC2a complex, which cleaves C5 to generate two different fragments, namely C5a and C5b [14]. The LP is very similar to the CP. The activated LP complex has an oligomer structure similar to the pentamolecular C1 complex [15] and is triggered by serine proteases associated with mannose-binding lectins (MBLs) and with ficolins, another family of lectins, which are able to recognize pathogens [16]. Upon activation by these signals, the enzymes of the complex mannan-binding lectin serine protease (MASP) 1 and 2 mediate the formation of the C3 convertase C4bC2a, which activates the same downstream pathways as occurs in CP [17]. In contrast to the classic and lectin pathways, the AP can autoactivate using a process termed “tickover” of C3 [18]. Initially, a spontaneously generated thioester- hydrolysed form of C3 (C3(H2O)) interacts with factor B and factor D to form the fluid phase alternative pathway C3 convertase C3(H2O)Bb [19]. C3(H2O)Bb cleaves native C3 to generate C3a and C3b, and the latter binds to its receptor on the lipid membrane. On the membrane, C3b combines with factor B, which is cleaved by factor D to form the alternative pathway C3 convertase C3bBb [14]. Properdin (P), a positive regulator of the complement system, stabilizes C3bBb, and the binding of additional C3b (amplification loop) to the existing alternative pathway C3 convertase generates a C5 convertase, thus leading to the production of C5a and C5b [14].
As it is a very complex system, various mechanisms can interfere with the complement cascade leading to over-activation and consequent neuronal damage and disease [20]. Dysregulated complement mechanisms, such as those involving C5 components, contribute to the initiation and progression of several neuropathies [21][22].
3. Peripheral Neuropathies and C5a/C5aR1 Axis
The term “neuropathy”, or also peripheral neuropathy (PN), refers to a group of conditions characterized by damage and loss of function of nerve cells in the brain or peripheral nervous system (PNS). The population prevalence is about 2400 per 100,000 rising with age to 8000 per 100,000 [23]. Although the damage occurs most frequently in the PNS, also brain injuries, such as stroke, can result in neuropathic symptoms [24]. Moreover, neuropathies can be expressions of neurodegenerative diseases [25], where the degeneration of sensory nerve fibres is due to a wide variety of insults, including diabetes, infectious diseases and nutritional deficiencies, and chemotherapy treatments [26]. Symptoms usually include numbness and paresthesia and are often accompanied by weakness and pain [27][28].
So far, progress in developing treatments for neuropathies has been frustratingly slow. In fact, despite the availability of therapies that can alleviate symptoms—as, for example, in the case of mild pain, which may be relieved by over the counter analgesics and topical patches—and can address conditions associated with PN [29][30][31][32], no treatments have been approved to date that directly modulate the underlying mechanisms of neuropathies. Current pharmacological therapies are only partially effective, and prolonged exposure to such agents can cause unwanted side effects. Consequently, there is an urgent need to identify and label specific molecular targets and to develop agents to treat pain by exploiting alternative biological pathways.
Over the last few years, evidence has indicated that C5a activation triggers a cascade of events that are involved in the pathophysiology of PN and in the genesis of painful states of neuro-inflammation [8]. C5a exerts its biological functions by binding two receptors, C5a receptor-like 1 (C5aR1, also referred to as CD88), a class A seven-transmembrane G-protein-coupled receptor (GPCR), and C5a receptor-like 2 (C5aR2, also known as C5L2 or GPR77) [33], a homolog of C5aR1, but which is not coupled to intracellular heterotrimeric G-proteins due to a mutation in G-protein recognition sequence. C5aR1 is expressed by a broad range of cell types, including all cells of myeloid origin (neutrophils, eosinophils, monocytes, macrophages, dendritic cells, mast cells), lymphocytes, and non-myeloid cells, such as lung, liver, kidney, skin, and central nervous system (CNS) cells [5][34]. C5aR2 is highly expressed in human tissues, such as bone marrow, spleen, and lung, as well as in immune cells, including most myeloid cells and specific T cell subsets [35]. C5aR1 is well-known for its pro-inflammatory effect [36]; conversely, the role of C5aR2 is poorly understood and still controversial [37]. Although C5aR2 can independently induce and modulate C5a biological functions through α-arrestin signalling, further investigations are needed to better understand its actual role [38].
By contrast, the C5a/C5aR1 axis triggers leukocyte recruitment and pro-inflammatory cytokines production, which drive inflammatory and neuropathic pain [39][40][41]. Up-regulated levels of C5a and C5aR were found in spinal cord microglia in animals subjected to spared nerve injury (SNI), a model of neuropathic pain [42], while local activation of C5aR1 was found to be implicated in the mechanical nociceptive sensitization in an in vivo model of postoperative pain [43]. In a similar model, PMX-53, a C5aR1 antagonist, decreased mechanical allodynia, oedema, and the levels of several inflammatory mediators present in incised skin [44]. Moreover, local pre-treatment of rats with PMX-53, attenuated mechanical hyperalgesia induced by zymosan, carrageenan, lipopolysaccharide, and ovalbumin, suggesting its role in the control of inflammatory pain [45]. In addition, oral administration of DF2593A, a non-competitive allosteric C5a inhibitor, effectively reduced mechanical hyperalgesia in a carrageenan and complete Freund’s adjuvant-induced inflammatory pain model. Furthermore, DF2593A reduced mechanical hypersensitivity in a model of neuropathic pain induced by SNI [40]. Notably, C5aR1 disruption in knock-out (KO) mice suppressed thermal hyperalgesia compared to wild-type (WT) mice and decreased mechanical sensitization after paw incision [40][41][43], suggesting a major involvement of C5a/C5aR1 axis in pain and inflammation after surgery.
In sum, the administration of C5aR1 antagonists produce analgesic effects in various models of inflammatory and neuropathic pain, highlighting the therapeutic potential of pharmacologically targeting the C5a/C5aR1 axis for chronic pain management.
4. Disease of the Peripheral Nervous System (PNS)
The role of C5a/C5aR axis activation in pain generation in neuropathies has been widely investigated in several pharmacological studies. The following paragraphs describe the more recent major findings in the field of PNS and discuss possible implications of the complement system in the pathogenesis of different neuropathic disorders.
4.1. Guillain-Barré Syndrome
GBS is a clinically heterogeneous spectrum of rare post-infectious neuropathies that usually occur in otherwise healthy patients and encompasses acute inflammatory demyelinating polyradiculoneuropathy (AIDP), acute motor axonal neuropathy (AMAN), acute motor-sensory axonal neuropathy (AMSAN), Miller–Fisher syndrome (MFS) and some other regional variants [46][47][48]. GBS is estimated to affect about 1 in 100,000 people each year and it can strike at any age and both sexes [49]. The exact cause of GBS is not known; it is characterized by symptoms that often affect the arms, breathing muscles, and even the face, reflecting widespread nerve damage. Several pathologic and etiologic subtypes of GBS exist, and in many cases it develops subsequently to minor infections but is not associated with other autoimmune or systemic disorders. Usually, GBS occurs after an infectious disease, during which antibodies that cross-react with gangliosides at nerve membranes-with anti-GQ1b ganglioside antibodies being the principal biomarkers of GBS [50]—are aberrantly generated and directed against the PNS, causing nerve damage or impairment of nerve conduction [51]. Anti-GQ1b ganglioside antibodies are principal biomarkers of GBS [50]. The concept of infection-triggered antibody cross-reactivity is well established in axonal GBS and this mechanism is suspected to play a key role in demyelinating GBS. Intravenous administration of immunoglobulins and plasma exchange are effective in treating GBS [52][53]; other therapeutic strategies have been tested in animal models, but their bench-to-bedside transfer is still lacking [54].
Inhibition of C5 complement component activation in experimental ex vivo and in vivo GBS models was extensively used to investigate the pathogenesis of GBS and to evaluate complement deposition in the nerve membrane [55][56][57][58][59]. Specifically, the complement inhibitor APT070 (Mirococept), which regulates C5 and C3 convertases, was shown to be efficacious in an anti-GQ1b-mediated mouse model of the GBS variant MFS, inhibiting the formation of MAC complexes and protecting nerve terminals [57]. Similarly, the anti-C5a monoclonal antibody eculizumab, which inhibits formation of C5a and C5b-9, was reported to prevent complement damage and respiratory paralysis in another severe in vivo mouse model of MFS generated via anti-GQ1b antibody and normal human serum injection as a complement source [58]. Together, these findings have raised the possibility of developing clinical trials using anti-C5a in GBS and in other antibody-mediated terminal motor neuropathies involving complement activation.
4.2. Chronic Inflammatory Demyelinating Polyradiculoneuropathy
CIDP is the most common chronic inflammatory neuropathy, and it is usually characterized by slow progressive, symmetric, proximal and distal paresis and sensory dysfunction. Symptoms develop in few months and the disease course can be either chronically progressive or relapsing with stepwise progression [60]. Prevalence is about 1 in 200,000 in children and 1–7 in 100,000 in adults, but it is recently accepted that the frequency is underestimated [61]. Although CIDP has been classified as an autoimmune disorder, in which an aberrant immune response is directed towards components of peripheral nerves causing segmental and multifocal demyelination, axonal degeneration and perivascular or endoneurial inflammatory infiltrates of macrophages and T cells, the exact mechanisms underlying the development of its immunopathology is still far from to be defined.
Individuals with CIDP lack a detectable antibody titer specific for major compact myelin proteins, thus suggesting that serum constituents, such as cytokines or components of the complement cascade, rather than myelin-directed antibodies might contribute to peripheral nerve injury [60][62][63]. Supporting the hypothesis that complement activation can be a potential pathogenic mechanism for this disease, complement component C3d deposition has been detected on the outer surface of PNS Schwann cells in biopsies from patients with CIDP [60][64][65]. Moreover, clinical studies demonstrated that CIDP patients have increased serum and cerebrospinal fluid levels of C5a [66], which is the result of the proinflammatory function of C3d aimed at recruiting myeloid cells, such as macrophages, to inflammation sites through complement receptors and inducing tissue injury through formation of the MAC. These findings suggest that systemic and local terminal complement activation is a characteristic feature of inflammatory demyelinating polyneuropathies and support a role of complement activation in the pathogenesis of CIDP.
4.3. Familial Amyloid Polyneuropathy
FAP, or transthyretin (TTR) amyloid polyneuropathy, is a progressive sensorimotor and autonomic neuropathy of adult onset, which is characterized by systemic accumulation of amyloid fibrils constituted of aberrant TTR protein [67]. The global prevalence is unknown, but in Japan it has been recently estimated to be around 1 person per million in the general population [68]. FAP is a heterogeneous disorder with a clinical presentation that varies based on the genotype and geographic origin [69][70]. To date, more than 40 TTR mutations have been identified and associated with different patterns of organ involvement, age of onset and disease progression [71][72]. The most common type of mutation is a substitution of valine for methionine at position 30 (ATTRV30M) [73]. The symptoms depend on the site of protein accumulation in the body and, although each TTR variant leads to a different phenotype, PN and cardiomyopathy are predominant hallmarks [74]. The disease usually worsens over 5 to 15 years, and often leads to death caused by heart failure due to TTR protein deposits. Liver transplantation is currently the only treatment for preventing synthesis of the amyloidogenic variants of TTR [75].
Nerve biopsies of individuals with amyloidogenic TTR revealed that in amyloid deposits, transthyretin is aggregated with several other proteins, such as apolipoprotein E, serum amyloid P, and complement C1q [76], suggesting a role for C1q in the pathogenesis of the disease. C1q protein has been shown to be involved also in other amyloidosis, such as Alzheimer’s disease, activating the complement pathway leading to neuronal loss [77]. It is speculated that the complement plays a dual role: although it is known that C1q is able to exert a neuroprotective function against toxic concentrations of soluble pre-amyloid aggregates [78][79], C5a is recognized as having a detrimental neuro-inflammatory effect [80]. However, the impact of C5aR/C5a axis activation in ATTRV30M amyloidosis remains to be clarified.
4.4. Chemotherapy-Induced Peripheral Neuropathy
CIPN is the most common neurologic complication of chemotherapy, often limiting the efficacy of cancer treatments [81]. Between 30% and 40% of patients receiving chemotherapy are reported to experience CIPN, and this number is expected to grow as more aggressive pharmacological agents emerge and survival rates increase [82]. The incidence of CIPN varies from 10% to 100%, depending upon the specific anticancer drug or drug combination administered and upon the dosing regimen [83]. CIPN causes pain, sensory loss and poor dexterity, with a significant impact on patient quality of life. When pain is too severe, a change in chemotherapy regimen may be required, with risk of reducing the therapeutic efficacy, or patients may choose to discontinue the treatment [84]. For example, both oxaliplatin and paclitaxel, two widely used chemotherapeutics, have been shown to cause neurotoxicity and alterations in sensory neurons, triggering CIPN [85][86].
Current CIPN management is far from satisfactory, and this is largely due to an inadequate understanding of the complexity of CIPN pathophysiology. The main neurobiological mechanisms involved in CIPN include impaired immune cell signalling and ion channel expression, neurotoxicity, mitochondrial dysfunction, and axon degeneration [87]. Emerging evidence suggests that the immune system and immune-mediated neuro-inflammation are crucial events in the development of CIPN [88]. In particular, a recent study reported that in paclitaxel-induced mechanical allodynia the complement cascade is reduced in C3 KO rats compared to WT animals, and that MAC is tightly involved in the damage of neuronal cells, suggesting that complement may be a novel target for the treatment of CIPN [89]. However, since C3 deficiency almost completely abolishes the release of C5a and MAC, which are crucial for the physiological protection against pathogens, and thus exposes the patient to an increased risk of infections and related side effects, the role of C5a-mediated signalling in CIPN models should be further investigated with the aim to develop more targeted treatment.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines9040399
References
- Nesargikar, P.; Spiller, B.; Chavez, R. The complement system: History, pathways, cascade and inhibitors. Eur. J. Microbiol. Immunol. 2012, 2, 103–111.
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262.
- Ricklin, D.; Lambris, J.D. Complement in immune and inflammatory disorders: Pathophysiological mechanisms. J. Immunol. 2013, 190, 3831–3838.
- Manthey, H.D.; Woodruff, T.M.; Taylor, S.M.; Monk, P.N. Complement component 5a (C5a). Int. J. Biochem. Cell Biol. 2009, 41, 2114–2117.
- Guo, R.F.; Ward, P.A. Role of C5a in inflammatory responses. Annu. Rev. Immunol. 2005, 23, 821–852.
- Heesterbeek, D.A.; Bardoel, B.W.; Parsons, E.S.; Bennett, I.; Ruyken, M.; Doorduijn, D.J.; Gorham, R.D., Jr.; Berends, E.T.; Pyne, A.L.; Hoogenboom, B.W.; et al. Bacterial killing by complement requires membrane attack complex formation via surface-bound C5 convertases. EMBO J. 2019, 38, e99852.
- Fritzinger, D.C.; Benjamin, D.E. The Complement System in Neuropathic and Postoperative Pain. Open Pain J. 2016, 9, 26–37.
- Quadros, A.U.; Cunha, T.M. C5a and pain development: An old molecule, a new target. Pharmacol. Res. 2016, 112, 58–67.
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257.
- Mamidi, S.; Hone, S.; Kirschfink, M. The complement system in cancer: Ambivalence between tumour destruction and promotion. Immunobiology 2017, 222, 45–54.
- Cedzynski, M.; Swierzko, A.S. Components of the Lectin Pathway of Complement in Haematologic Malignancies. Cancers 2020, 12, 1792.
- Mortensen, S.A.; Sander, B.; Jensen, R.K.; Pedersen, J.S.; Golas, M.M.; Thiel, S.; Andersen, G.R. Models of the complement C1 complex. Proc. Natl. Acad. Sci. USA 2018, 115, E3866.
- Gaboriaud, C.; Thielens, N.M.; Gregory, L.A.; Rossi, V.; Fontecilla-Camps, J.C.; Arlaud, G.J. Structure and activation of the C1 complex of complement: Unraveling the puzzle. Trends Immunol. 2004, 25, 368–373.
- Zwarthoff, S.A.; Berends, E.T.M.; Mol, S.; Ruyken, M.; Aerts, P.C.; Jozsi, M.; de Haas, C.J.C.; Rooijakkers, S.H.M.; Gorham, R.D., Jr. Functional Characterization of Alternative and Classical Pathway C3/C5 Convertase Activity and Inhibition Using Purified Models. Front. Immunol. 2018, 9, 1691.
- Kjaer, T.R.; Jensen, L.; Hansen, A.; Dani, R.; Jensenius, J.C.; Dobo, J.; Gal, P.; Thiel, S. Oligomerization of Mannan-binding Lectin Dictates Binding Properties and Complement Activation. Scand. J. Immunol. 2016, 84, 12–19.
- Takahashi, M.; Iwaki, D.; Kanno, K.; Ishida, Y.; Xiong, J.; Matsushita, M.; Endo, Y.; Miura, S.; Ishii, N.; Sugamura, K.; et al. Mannose-binding lectin (MBL)-associated serine protease (MASP)-1 contributes to activation of the lectin complement pathway. J. Immunol. 2008, 180, 6132–6138.
- Mortensen, S.; Kidmose, R.T.; Petersen, S.V.; Szilagyi, A.; Prohaszka, Z.; Andersen, G.R. Structural Basis for the Function of Complement Component C4 within the Classical and Lectin Pathways of Complement. J. Immunol. 2015, 194, 5488–5496.
- Thurman, J.M.; Holers, V.M. The central role of the alternative complement pathway in human disease. J. Immunol. 2006, 176, 1305–1310.
- Chen, Z.A.; Pellarin, R.; Fischer, L.; Sali, A.; Nilges, M.; Barlow, P.N.; Rappsilber, J. Structure of Complement C3(H2O) Revealed By Quantitative Cross-Linking/Mass Spectrometry And Modeling. Mol. Cell Proteom. 2016, 15, 2730–2743.
- Hernandez, M.X.; Namiranian, P.; Nguyen, E.; Fonseca, M.I.; Tenner, A.J. C5a Increases the Injury to Primary Neurons Elicited by Fibrillar Amyloid Beta. ASN Neuro 2017, 9, 1759091416687871.
- Ramaglia, V.; Daha, M.R.; Baas, F. The complement system in the peripheral nerve: Friend or foe? Mol. Immunol. 2008, 45, 3865–3877.
- Dalakas, M.C.; Alexopoulos, H.; Spaeth, P.J. Complement in neurological disorders and emerging complement-targeted therapeutics. Nat. Rev. Neurol. 2020, 16, 601–617.
- Hughes, R.A. Peripheral neuropathy. BMJ 2002, 324, 466–469.
- Costigan, M.; Scholz, J.; Woolf, C.J. Neuropathic pain: A maladaptive response of the nervous system to damage. Annu. Rev. Neurosci. 2009, 32, 1–32.
- Reichling, D.B.; Levine, J.D. Pain and death: Neurodegenerative disease mechanisms in the nociceptor. Ann. Neurol. 2011, 69, 13–21.
- Jin, H.W.; Flatters, S.J.; Xiao, W.H.; Mulhern, H.L.; Bennett, G.J. Prevention of paclitaxel-evoked painful peripheral neuropathy by acetyl-L-carnitine: Effects on axonal mitochondria, sensory nerve fiber terminal arbors, and cutaneous Langerhans cells. Exp. Neurol. 2008, 210, 229–237.
- Head, K.A. Peripheral neuropathy: Pathogenic mechanisms and alternative therapies. Altern. Med. Rev. 2006, 11, 294–329.
- Hanewinckel, R.; Ikram, M.A.; Van Doorn, P.A. Peripheral neuropathies. Handb. Clin. Neurol. 2016, 138, 263–282.
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 170002.
- Girach, A.; Julian, T.H.; Varrassi, G.; Paladini, A.; Vadalouka, A.; Zis, P. Quality of Life in Painful Peripheral Neuropathies: A Systematic Review. Pain Res. Manag. 2019, 2019, 2091960.
- Liampas, A.; Rekatsina, M.; Vadalouca, A.; Paladini, A.; Varrassi, G.; Zis, P. Non-Pharmacological Management of Painful Peripheral Neuropathies: A Systematic Review. Adv. Ther. 2020, 37, 4096–4106.
- Liampas, A.; Rekatsina, M.; Vadalouca, A.; Paladini, A.; Varrassi, G.; Zis, P. Pharmacological Management of Painful Peripheral Neuropathies: A Systematic Review. Pain Ther. 2020, 1–14.
- Nabizadeh, J.A.; Manthey, H.D.; Panagides, N.; Steyn, F.J.; Lee, J.D.; Li, X.X.; Akhir, F.N.M.; Chen, W.; Boyle, G.M.; Taylor, S.M.; et al. C5a receptors C5aR1 and C5aR2 mediate opposing pathologies in a mouse model of melanoma. FASEB J. 2019, 33, 11060–11071.
- Allegretti, M.; Moriconi, A.; Beccari, A.R.; Di Bitondo, R.; Bizzarri, C.; Bertini, R.; Colotta, F. Targeting C5a: Recent advances in drug discovery. Curr. Med. Chem. 2005, 12, 217–236.
- Li, X.X.; Lee, J.D.; Kemper, C.; Woodruff, T.M. The Complement Receptor C5aR2: A Powerful Modulator of Innate and Adaptive Immunity. J. Immunol. 2019, 202, 3339–3348.
- Peng, Q.; Li, K.; Sacks, S.H.; Zhou, W. The role of anaphylatoxins C3a and C5a in regulating innate and adaptive immune responses. Inflamm. Allergy Drug Targets 2009, 8, 236–246.
- Zhang, T.; Garstka, M.A.; Li, K. The Controversial C5a Receptor C5aR2: Its Role in Health and Disease. J. Immunol. Res. 2017, 2017, 8193932.
- Croker, D.E.; Monk, P.N.; Halai, R.; Kaeslin, G.; Schofield, Z.; Wu, M.C.; Clark, R.J.; Blaskovich, M.A.; Morikis, D.; Floudas, C.A.; et al. Discovery of functionally selective C5aR2 ligands: Novel modulators of C5a signalling. Immunol. Cell Biol. 2016, 94, 787–795.
- Wiese, A.V.; Ender, F.; Quell, K.M.; Antoniou, K.; Vollbrandt, T.; Konig, P.; Kohl, J.; Laumonnier, Y. The C5a/C5aR1 axis controls the development of experimental allergic asthma independent of LysM-expressing pulmonary immune cells. PLoS ONE 2017, 12, e0184956.
- Moriconi, A.; Cunha, T.M.; Souza, G.R.; Lopes, A.H.; Cunha, F.Q.; Carneiro, V.L.; Pinto, L.G.; Brandolini, L.; Aramini, A.; Bizzarri, C.; et al. Targeting the minor pocket of C5aR for the rational design of an oral allosteric inhibitor for inflammatory and neuropathic pain relief. Proc. Natl. Acad. Sci. USA 2014, 111, 16937–16942.
- Shutov, L.P.; Warwick, C.A.; Shi, X.; Gnanasekaran, A.; Shepherd, A.J.; Mohapatra, D.P.; Woodruff, T.M.; Clark, J.D.; Usachev, Y.M. The Complement System Component C5a Produces Thermal Hyperalgesia via Macrophage-to-Nociceptor Signaling That Requires NGF and TRPV1. J. Neurosci. 2016, 36, 5055–5070.
- Griffin, R.S.; Costigan, M.; Brenner, G.J.; Ma, C.H.; Scholz, J.; Moss, A.; Allchorne, A.J.; Stahl, G.L.; Woolf, C.J. Complement induction in spinal cord microglia results in anaphylatoxin C5a-mediated pain hypersensitivity. J. Neurosci. 2007, 27, 8699–8708.
- Liang, D.Y.; Li, X.; Shi, X.; Sun, Y.; Sahbaie, P.; Li, W.W.; Clark, J.D. The complement component C5a receptor mediates pain and inflammation in a postsurgical pain model. Pain 2012, 153, 366–372.
- Clark, J.D.; Qiao, Y.; Li, X.; Shi, X.; Angst, M.S.; Yeomans, D.C. Blockade of the complement C5a receptor reduces incisional allodynia, edema, and cytokine expression. Anesthesiology 2006, 104, 1274–1282.
- Ting, E.; Guerrero, A.T.; Cunha, T.M.; Verri, W.A., Jr.; Taylor, S.M.; Woodruff, T.M.; Cunha, F.Q.; Ferreira, S.H. Role of complement C5a in mechanical inflammatory hypernociception: Potential use of C5a receptor antagonists to control inflammatory pain. Br. J. Pharmacol. 2008, 153, 1043–1053.
- Hafer-Macko, C.; Hsieh, S.T.; Li, C.Y.; Ho, T.W.; Sheikh, K.; Cornblath, D.R.; McKhann, G.M.; Asbury, A.K.; Griffin, J.W. Acute motor axonal neuropathy: An antibody-mediated attack on axolemma. Ann. Neurol. 1996, 40, 635–644.
- Kuwabara, S.; Yuki, N. Axonal Guillain-Barre syndrome: Concepts and controversies. Lancet Neurol. 2013, 12, 1180–1188.
- Jasti, A.K.; Selmi, C.; Sarmiento-Monroy, J.C.; Vega, D.A.; Anaya, J.M.; Gershwin, M.E. Guillain-Barre syndrome: Causes, immunopathogenic mechanisms and treatment. Expert Rev. Clin. Immunol. 2016, 12, 1175–1189.
- McGrogan, A.; Madle, G.C.; Seaman, H.E.; de Vries, C.S. The epidemiology of Guillain-Barre syndrome worldwide. A systematic literature review. Neuroepidemiology 2009, 32, 150–163.
- Jacobs, B.C.; O’Hanlon, G.M.; Bullens, R.W.; Veitch, J.; Plomp, J.J.; Willison, H.J. Immunoglobulins inhibit pathophysiological effects of anti-GQ1b-positive sera at motor nerve terminals through inhibition of antibody binding. Brain 2003, 126, 2220–2234.
- Fokke, C.; van den Berg, B.; Drenthen, J.; Walgaard, C.; van Doorn, P.A.; Jacobs, B.C. Diagnosis of Guillain-Barre syndrome and validation of Brighton criteria. Brain 2014, 137, 33–43.
- Schaller, B.; Radziwill, A.J.; Steck, A.J. Successful treatment of Guillain-Barre syndrome with combined administration of interferon-beta-1a and intravenous immunoglobulin. Eur. Neurol. 2001, 46, 167–168.
- Raphael, J.C.; Chevret, S.; Hughes, R.A.; Annane, D. Plasma exchange for Guillain-Barre syndrome. Cochrane Database Syst. Rev. 2002, 7, CD001798.
- Meyer zu Horste, G.; Hartung, H.P.; Kieseier, B.C. From bench to bedside--experimental rationale for immune-specific therapies in the inflamed peripheral nerve. Nat. Clin. Pract. Neurol. 2007, 3, 198–211.
- O’Hanlon, G.M.; Plomp, J.J.; Chakrabarti, M.; Morrison, I.; Wagner, E.R.; Goodyear, C.S.; Yin, X.; Trapp, B.D.; Conner, J.; Molenaar, P.C.; et al. Anti-GQ1b ganglioside antibodies mediate complement-dependent destruction of the motor nerve terminal. Brain 2001, 124, 893–906.
- Halstead, S.K.; O’Hanlon, G.M.; Humphreys, P.D.; Morrison, D.B.; Morgan, B.P.; Todd, A.J.; Plomp, J.J.; Willison, H.J. Anti-disialoside antibodies kill perisynaptic Schwann cells and damage motor nerve terminals via membrane attack complex in a murine model of neuropathy. Brain 2004, 127, 2109–2123.
- Halstead, S.K.; Humphreys, P.D.; Goodfellow, J.A.; Wagner, E.R.; Smith, R.A.; Willison, H.J. Complement inhibition abrogates nerve terminal injury in Miller Fisher syndrome. Ann. Neurol. 2005, 58, 203–210.
- Halstead, S.K.; Zitman, F.M.; Humphreys, P.D.; Greenshields, K.; Verschuuren, J.J.; Jacobs, B.C.; Rother, R.P.; Plomp, J.J.; Willison, H.J. Eculizumab prevents anti-ganglioside antibody-mediated neuropathy in a murine model. Brain 2008, 131, 1197–1208.
- McGonigal, R.; Cunningham, M.E.; Yao, D.; Barrie, J.A.; Sankaranarayanan, S.; Fewou, S.N.; Furukawa, K.; Yednock, T.A.; Willison, H.J. C1q-targeted inhibition of the classical complement pathway prevents injury in a novel mouse model of acute motor axonal neuropathy. Acta Neuropathol. Commun. 2016, 4, 1–16.
- Dalakas, M.C. Medscape. Advances in the diagnosis, pathogenesis and treatment of CIDP. Nat. Rev. Neurol. 2011, 7, 507–517.
- Broers, M.C.; Bunschoten, C.; Nieboer, D.; Lingsma, H.F.; Jacobs, B.C. Incidence and Prevalence of Chronic Inflammatory Demyelinating Polyradiculoneuropathy: A Systematic Review and Meta-Analysis. Neuroepidemiology 2019, 52, 161–172.
- Koller, H.; Kieseier, B.C.; Jander, S.; Hartung, H.P. Chronic inflammatory demyelinating polyneuropathy. N. Engl. J. Med. 2005, 352, 1343–1356.
- Mathey, E.K.; Park, S.B.; Hughes, R.A.; Pollard, J.D.; Armati, P.J.; Barnett, M.H.; Taylor, B.V.; Dyck, P.J.; Kiernan, M.C.; Lin, C.S. Chronic inflammatory demyelinating polyradiculoneuropathy: From pathology to phenotype. J. Neurol. Neurosurg. Psychiatry 2015, 86, 973–985.
- Dalakas, M.C.; Engel, W.K. Immunoglobulin and complement deposits in nerves of patients with chronic relapsing polyneuropathy. Arch. Neurol. 1980, 37, 637–640.
- Hays, A.P.; Lee, S.S.; Latov, N. Immune reactive C3d on the surface of myelin sheaths in neuropathy. J. Neuroimmunol. 1988, 18, 231–244.
- Quast, I.; Keller, C.W.; Hiepe, F.; Tackenberg, B.; Lunemann, J.D. Terminal complement activation is increased and associated with disease severity in CIDP. Ann. Clin. Transl. Neurol. 2016, 3, 730–735.
- Cakar, A.; Durmus-Tekce, H.; Parman, Y. Familial Amyloid Polyneuropathy. Arch. Neurol. 2019, 56, 150.
- Kato-Motozaki, Y.; Ono, K.; Shima, K.; Morinaga, A.; Machiya, T.; Nozaki, I.; Shibata-Hamaguchi, A.; Furukawa, Y.; Yanase, D.; Ishida, C.; et al. Epidemiology of familial amyloid polyneuropathy in Japan: Identification of a novel endemic focus. J. Neurol. Sci. 2008, 270, 133–140.
- Ando, Y.; Nakamura, M.; Araki, S. Transthyretin-related familial amyloidotic polyneuropathy. Arch. Neurol. 2005, 62, 1057–1062.
- Plante-Bordeneuve, V.; Said, G. Familial amyloid polyneuropathy. Lancet Neurol. 2011, 10, 1086–1097.
- Rowczenio, D.; Quarta, C.C.; Fontana, M.; Whelan, C.J.; Martinez-Naharro, A.; Trojer, H.; Baginska, A.; Ferguson, S.M.; Gilbertson, J.; Rezk, T.; et al. Analysis of the TTR gene in the investigation of amyloidosis: A 25-year single UK center experience. Hum. Mutat. 2019, 40, 90–96.
- Manganelli, F.; Fabrizi, G.M.; Luigetti, M.; Mandich, P.; Mazzeo, A.; Pareyson, D. Hereditary transthyretin amyloidosis overview. Neurol. Sci. 2020.
- Ohmori, H.; Ando, Y.; Makita, Y.; Onouchi, Y.; Nakajima, T.; Saraiva, M.J.; Terazaki, H.; Suhr, O.; Sobue, G.; Nakamura, M.; et al. Common origin of the Val30Met mutation responsible for the amyloidogenic transthyretin type of familial amyloidotic polyneuropathy. J. Med. Genet. 2004, 41, e51.
- Ando, Y.; Coelho, T.; Berk, J.L.; Cruz, M.W.; Ericzon, B.G.; Ikeda, S.; Lewis, W.D.; Obici, L.; Plante-Bordeneuve, V.; Rapezzi, C.; et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J. Rare Dis. 2013, 8, 31.
- Benson, M.D. Liver transplantation and transthyretin amyloidosis. Muscle Nerve 2013, 47, 157–162.
- Hafer-Macko, C.E.; Dyck, P.J.; Koski, C.L. Complement activation in acquired and hereditary amyloid neuropathy. J. Peripher. Nerv. Syst. 2000, 5, 131–139.
- Fonseca, M.I.; Zhou, J.; Botto, M.; Tenner, A.J. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J. Neurosci. 2004, 24, 6457–6465.
- Pisalyaput, K.; Tenner, A.J. Complement component C1q inhibits beta-amyloid- and serum amyloid P-induced neurotoxicity via caspase- and calpain-independent mechanisms. J. Neurochem. 2008, 104, 696–707.
- Galvan, M.D.; Foreman, D.B.; Zeng, E.; Tan, J.C.; Bohlson, S.S. Complement component C1q regulates macrophage expression of Mer tyrosine kinase to promote clearance of apoptotic cells. J. Immunol. 2012, 188, 3716–3723.
- Fonseca, M.I.; Chu, S.H.; Berci, A.M.; Benoit, M.E.; Peters, D.G.; Kimura, Y.; Tenner, A.J. Contribution of complement activation pathways to neuropathology differs among mouse models of Alzheimer’s disease. J. Neuroinflamm. 2011, 8, 1–12.
- Quasthoff, S.; Hartung, H.P. Chemotherapy-induced peripheral neuropathy. J. Neurol. 2002, 249, 9–17.
- Pike, C.T.; Birnbaum, H.G.; Muehlenbein, C.E.; Pohl, G.M.; Natale, R.B. Healthcare costs and workloss burden of patients with chemotherapy-associated peripheral neuropathy in breast, ovarian, head and neck, and nonsmall cell lung cancer. Chemother. Res. Pract. 2012, 2012, 913848.
- Balayssac, D.; Ferrier, J.; Descoeur, J.; Ling, B.; Pezet, D.; Eschalier, A.; Authier, N. Chemotherapy-induced peripheral neuropathies: From clinical relevance to preclinical evidence. Expert Opin. Drug Saf. 2011, 10, 407–417.
- Kolb, N.A.; Smith, A.G.; Singleton, J.R.; Beck, S.L.; Stoddard, G.J.; Brown, S.; Mooney, K. The Association of Chemotherapy-Induced Peripheral Neuropathy Symptoms and the Risk of Falling. JAMA Neurol. 2016, 73, 860–866.
- Salat, K. Chemotherapy-induced peripheral neuropathy: Part 1-current state of knowledge and perspectives for pharmacotherapy. Pharmacol. Rep. 2020, 72, 486–507.
- Salat, K. Chemotherapy-induced peripheral neuropathy-part 2: Focus on the prevention of oxaliplatin-induced neurotoxicity. Pharmacol. Rep. 2020, 72, 508–527.
- Lees, J.G.; Makker, P.G.; Tonkin, R.S.; Abdulla, M.; Park, S.B.; Goldstein, D.; Moalem-Taylor, G. Immune-mediated processes implicated in chemotherapy-induced peripheral neuropathy. Eur. J. Cancer 2017, 73, 22–29.
- Brandolini, L.; d’Angelo, M.; Antonosante, A.; Allegretti, M.; Cimini, A. Chemokine Signaling in Chemotherapy-Induced Neuropathic Pain. Int. J. Mol. Sci. 2019, 20, 2904.
- Xu, J.; Zhang, L.; Xie, M.; Li, Y.; Huang, P.; Saunders, T.L.; Fox, D.A.; Rosenquist, R.; Lin, F. Role of Complement in a Rat Model of Paclitaxel-Induced Peripheral Neuropathy. J. Immunol. 2018, 200, 4094–4101.