Ca2+ is pivotal intracellular messenger that coordinates multiple cell functions such as fertilization, growth, differentiation, and viability. Intracellular Ca2+ signaling is regulated by both extracellular Ca2+ entry and Ca2+ release from intracellular stores. Apart from working as the cellular recycling center, the lysosome has been increasingly recognized as a significant intracellular Ca2+ store that provides Ca2+ to regulate many cellular processes. The lysosome also talks to other organelles by releasing and taking up Ca2+. In lysosomal Ca2+-dependent processes, autophagy is particularly important, because it has been implicated in many human diseases including cancer.
- lysosome
- ion channel
- calcium
- autophagy
- cancer
1. Introduction
The lysosome is an acidic single-membrane organelle first discovered in 1955 by Christian de Duve while investigating the mechanism of action of insulin [1][2][3]. It contains more than 60 hydrolytic enzymes (e.g., nucleases, glycosidase, phosphatases, sulfatases, lipases, and proteases), which are active at the luminal acidic environment (pH 4.5–5.0) established by the vacuolar-ATPase (V-ATPase) proton pump [4][5]. Since its discovery, the lysosome has mainly been considered to be the center of waste disposal, which digests unwanted macromolecules, damaged and senescent organelles, microbes, and other particles delivered via endocytosis, autophagy, and phagocytosis [4][6][7][8]. Once degraded, some breakdown products such as free fatty acids, amino acid, monosaccharides and nucleotides are transported back to the cytosol via specific exporters in the lysosome membrane for reutilization in anabolic processes [9][10]. Lysosomes also contain more than 60 membrane proteins that are implicated in the maintenance of the lumen homeostasis, especially ionic homeostasis and membrane potential, in the control of molecular export across the lysosomal membrane, and in lysosomal membrane trafficking (i.e., fusion and fission). The functions of lysosomes in material degradation, catabolite export, or trafficking are key to maintaining cellular homeostasis, the perturbations of which often lead to lysosomal storage diseases (LSDs) [11].
Recent studies have shown that the lysosome is not only the terminal degradative compartment, but also a multifunctional signaling hub that integrates the cell’s responses to nutrient status, growth factors, and hormones. Noticeably, in order to adapt to changes in cellular environment, the lysosome has a nutrient-sensing mechanism involving mammalian/mechanistic target of rapamycin complex 1 (mTORC1) and transcription factor EB (TFEB) [12][13][14]. mTORC1 is capable of sensing a myriad of nutrient and energy cues, phosphorylating numerous cell growth-related substrates including TFEB, and thus governing the balance between catabolic and anabolic metabolic pathways in the cell [15]. TFEB can bind to a palindromic 10 bp nucleotide motif, named the coordinated lysosomal expression, and regulation (CLEAR) element, and activate the transcription of many genes encoding lysosomal proteins and autophagy-related proteins [16][17]. Under nutrient sufficient conditions, TFEB is sequestered away from the nucleus due to being phosphorylated by mTORC1. Conversely, under starvation, TFEB becomes dephosphorylated due to a reduction in mTORC1 and an activation of calcineurin (CaN), a Ca2+ and calmodulin (CaM) dependent serine/threonine protein phosphatase, and translocates to the nucleus to promote the transcription of the CLEAR element, which subsequently promotes autophagy-lysosome pathway as well as exocytosis and phagocytosis [8][18][19][20].
Autophagy is an evolutionarily conserved cellular degradative process that is induced by nutrient and energy starvation. It is a fundamental cellular program for cells to maintain intracellular energy and nutrient homeostasis and to protect cells against stress. During autophagy, intracellular components such as macromolecules and unwanted organelles are engulfed in autophagosomes that then fuse with lysosomes to form autolysosomes for degradation [21]. Lysosomal Ca2+ plays a key role in autophagy. For example, Transient Receptor Potential Mucolipin 1 (TRPML1, encoded by MCOLN1 gene), an important Ca2+ channel in the lysosome, governs autophagy through regulating both mTORC1 [22][23] and TFEB [24][25]. Because autophagy has an essential role in cellular homeostasis, it is implicated in various physiological processes and human diseases. Among them, the roles of autophagy in cancer have been extensively studied.
2. Lysosomal Ca2+ Homeostasis
The lysosome is a significant intracellular Ca2+ store, which coordinates cellular adaptive responses [12][15][26][27][28][29][30]. In the early 1990s, nicotinic acid adenine dinucleotide phosphate (NAADP) was discovered to be a potent mobilizer of Ca2+ from stores separated from those sensitive to inositol 1,4,5-trisphosphate (IP3) and cyclic ADP-ribose (cADPR) [31]. It was later shown that the NAADP-sensitive Ca2+ store is the functional equivalent of the lysosomal system, suggesting the lysosome may function as a Ca2+ store [32]. Indeed, in the same time, the lysosomal Ca2+ concentration was estimated to be ~0.5 mM, approximately 5000-fold higher than the cytosolic Ca2+ concentration (∼100 nM) [33][34][35]. This Ca2+ gradient across the lysosomal membrane is thought to be established by an unidentified Ca2+/H+ exchanger or Ca2+ transporter [36][37]. Multiple Ca2+ sensors including CaM, apoptosis linked gene 2 (ALG-2), and synaptotagmin 7 (Syt 7) associated with lysosomes have also been identified [22][33][38][39][40][41]. This further increases the diversity of lysosomal Ca2+-dependent processes. Recently, several groups have made new discoveries elucidating the molecular machinery underlying lysosomal Ca2+ release, Ca2+ signaling, and Ca2+ uptake (Figure 1).
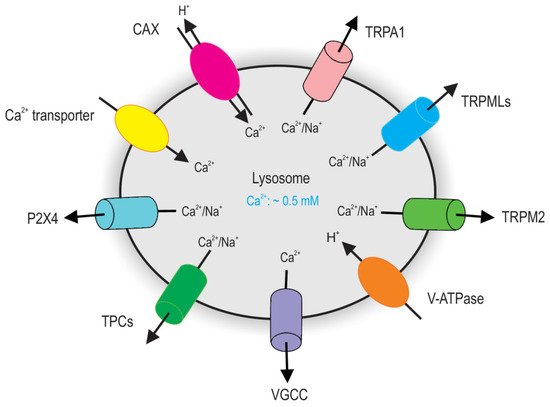
Figure 1. Major Ca2+ releasing channels and transporters on lysosomes. TRPML channels and TPC channels are major groups of Ca2+ channels on lysosomes that have been definitely defined by endolysosome-patch-clamp. Given the topology of the TRPML proteins at the endolysosomal membrane and the electrical properties of the endolysosome, TRPML opening leads to Ca2+ and Na+ release from the endolysosome to the cytosol. Activation of TPCs release lysosomal Na+ and Ca2+. Lysosomes accumulate Ca2+ using a putative Ca2+ Transporter or Ca2+/H+ exchanger (CAX).
3. Autophagy
Autophagy is a self-eating process that is important for balancing sources of energy at critical times in development and in response to nutrient stress. The cell uses an autophagy pathway to degrade and recycle cytoplasmic constituents such as protein aggregates, lipids, and complete organelles for cell survival. It is especially important in postmitotic cells, such as muscles and neurons, where accumulation of aggregated proteins and damaged organelles often results in cell death [42][43][44]. Indeed, suppression of autophagy causes compromised neuron and muscle differentiation [45][46][47] as well as neurodegeneration [42][43][44] and myofiber degeneration [48][49].
In most cells, autophagy is kept at a low level under nutrient rich condition. However, stressful conditions, such as nutritional deprivation, oxidative stress, Ca2+ overload, pathogen infection, and other diseases, activate autophagy. By upregulating autophagy under such conditions, cells degrade macromolecules into their building blocks for reutilization, thereby adapting to extreme conditions and maintaining cellular homeostasis.
There are three types of autophagy: macroautophagy [50][51], microautophagy [50][52], and chaperone-mediated autophagy (CMA) [53][54]. Macroautophagy is the most common form of autophagy, which is characterized by the formation of a typical double-membrane cistern (so called phagophore) that extends and engulfs part of the cytoplasm to form a whole vesicle (so called autophagosome). The autophagosome ultimately fuses with a lysosome to form an autolysosome [55][56][57]. In this review, we focus on macroautophagy (hereafter referred to as autophagy).
The process of autophagy is controlled by multiple complexes of proteins encoded by evolutionarily conserved, autophagy-related (ATG) genes, which were originally identified in yeast. The products of these ATG genes, together with other autophagy-related factors, regulate autophagosome formation, tethering, and fusion with lysosomes [58][59][60][61]. Autophagy is also regulated by some non-ATG proteins. For example, in the presence of nutrients, ATG1/ULK1 is phosphorylated by mTORC1 [62], thereby inhibiting autophagy initiation [63]. mTORC1 can also phosphorylate and inactivate TFEB, repressing autophagy [13][64]. In the absence of nutrients, TRPML1-metiated lysosomal Ca2+ release activates CaN, which further causes TFEB dephosphorylation and nuclear translocation, thereby promoting autophagy [25].
Studies of mammalian systems have highlighted many important roles of autophagy in health and diseases including cell growth [65] and differentiation [47], LSDs [66][67], neurodegenerative diseases [43][44], bacterial infections [68], and cancers [69][70].
4. Lysosomal Ca2+ in Autophagy
4.1. TRPML Channels in Autophagy
It is widely accepted that Ca2+ can regulate autophagy, while mechanisms differ depending on the conditions. The interplay between lysosomal Ca2+ signal and autophagy has also been reported. In line with this, several lysosomal Ca2+-permeable channels have been suggested to regulate autophagy [71][72][73].
As a key Ca2+ release channel in the lysosomal membrane, TRPML1 deficiency leads to defective autophagy including accumulation of autophagosomes and aggregation of p62 proteins [74][75][76][77]. Growing evidence suggests that TRPML1 plays multifaceted roles in autophagy. Under normal conditions, mTORC1 phosphorylates and inhibits both TRPML1 and TFEB. Cellular stress activates TRPML1 due to mTORC1 inhibition. This further activates downstream pathways including (1) CaM/CaMKKβ/AMPK-dependent autophagosome formation [78], (2) ALG-2-dependent lysosome centripetal movement to promote autophagosome–lysosome fusion [79], (3) proteolytic degradation in autolysosomes [80], (4) Syt7-dependent lysosomal exocytosis to remove cellular garbage [79][81], (5) CaM-dependent mTORC1 reactivation to prevent cell death by increasing protein synthesis and promote lysosome reformation [22][23], and (6) CaN/TFEB activation to continuously supply lysosome and autophagy proteins [24][25][82] (Figure 3). Because TRPML1 is also a target of TFEB, a positive feedback is established to largely potentiate autophagy during stress [24][25]. Thus, TRPML1 is involved in several steps of autophagy including autophagosome formation, autophagosome maturation, autolysosome degradation, and autophagic lysosome reformation.
In addition to TRPML1, TRPML3 also takes part in autophagy regulation. TRPML3 has been found in the plasma membrane and multiple intracellular compartments, including autophagosomes, early endosomes, late endosomes, and lysosomes. The multiple compartmental localization of TRPML3 suggests that TRPML3 is dynamically expressed in different compartments and plays a role in membrane trafficking. Indeed, TRPML3 is accumulated in the plasma membrane upon inhibition of endocytosis and is recruited to autophagosomes upon induction of autophagy, thereby regulating endocytosis and autophagy [83]. Specifically for autophagy, TRPML3 overexpression increases while its knock-down or expression of the channel-dead dominant negative TRPML3 reduces autophagy [83]. Mechanistically, emerging evidence suggests that palmitoylation at its C-terminal region is required for TRPML3′s function in autophagosome formation, potentially by controlling its trafficking to autophagic structures [84], where TRPML3 may promote autophagosome maturation by providing Ca2+ in the fusion process through a specific interaction with GATE16, a mammalian ATG8 homologue [85].
4.2. TPC Channels in Autophagy
The role of TPCs in autophagy has been conflicting. Pereira et al. [86] showed that in astrocytes, NAADP and TPC2 overexpression increased the levels of autophagy markers, LC3 and beclin-1, and NAADP-mediated increases in LC3II levels were reduced in cells expressing a dominant–negative TPC2 construct. In the meantime, Leucine-rich repeat kinase 2 (LRRK2), an important regulator of autophagy involved in late-onset familial Parkinson’s disease (PD) [87], activated the CaMKKβ)/AMPK pathway, which was followed by a persistent increase in autophagosome formation. These effects were mimicked by the lysosomal Ca2+-mobilizing messenger NAADP and reversed by an NAADP receptor antagonist or expression of dominant–negative receptor constructs, suggesting that TPC2-mediated lysosomal Ca2+ release may promote autophagy [88]. However, skeletal muscles from animals lacking TPC2 displayed an enhanced autophagy flux [89]. In addition, loss of TPCs did not appear to have gross defects in autophagy in the liver, heart, and macrophages [90]. There, the role of TPC2 in autophagy may be dependent on the conditions. Interestingly, Cang et al. suggested that ATP/mTOR phosphorylates and inhibits TPCs, thereby acting as a nutrient sensor to detect nutrient status in response to intracellular ATP and mTOR levels [90]. In contrast, in skeletal muscle, the loss of TPC2 leads to a reduced mTOR [89]. It seems that TPC2 and mTOR form a feedback regulatory loop in response to nutrient status.
4.3. Other Channels in Autophagy
Although most voltage gated Ca2+ channels (VGCCs) are found in the plasma membrane of excitable cells, P/Q-type VGCCs are recently reported to be expressed in lysosomes of both mice and fruit flies. Loss of VGCC leads to defects in autophagosome–lysosome fusion, indicating an important role of Ca2+ flux through this channel in autophagy [91].
5. Lysosomal Ca2+, Autophagy, and Cancer
An increasing number of tumorigenic pathways have been associated with an altered expression level or abnormal activation of Ca2+ regulatory membrane proteins including Ca2+ channels, transporters, or Ca2+-ATPases [92][93][94][95][96][97]. Abnormal autophagy has also been implicated in cancer development. It protects against the initiation of carcinogenesis, but also has a role enabling the survival of cells in solid tumors where nutrients are limited [98][99][100][101][102]. Given that lysosomal Ca2+ channels play an important role in autophagy, the role of lysosomal Ca2+ channels in cancer development has attracted great attention in recent years [24][25][103][104][105]. It is believed that impaired lysosomal Ca2+ signaling is a culprit in malignant tumor development [74]. Indeed, emerging evidence has demonstrated that lysosomal Ca2+ signaling underlies several cancer hallmarks involving proliferation, metastasis, and angiogenesis and contributes to multidrug resistance in cancer therapy [74][106][107][108][109][110][111][112][113][114][115][116]. Here we discuss the roles of the two major Ca2+ permeable channels, TRPMLs and TPCs, in cancer.
This entry is adapted from the peer-reviewed paper 10.3390/cancers13061299
References
- De Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 1955, 60, 604–617.
- Novikoff, A.B.; Beaufay, H.; De Duve, C. Electron microscopy of lysosomerich fractions from rat liver. J. Biophys. Biochem. Cytol. 1956, 2, 179–184.
- Sabatini, D.D.; Adesnik, M. Christian de Duve: Explorer of the cell who discovered new organelles by using a centrifuge. Proc. Natl. Acad. Sci. USA 2013, 110, 13234–13235.
- Luzio, J.P.; Hackmann, Y.; Dieckmann, N.M.G.; Griffiths, G.M. The Biogenesis of Lysosomes and Lysosome-Related Organelles. Cold Spring Harb. Perspect. Biol. 2014, 6, a016840.
- Chakraborty, K.; Leung, K.; Krishnan, Y. High lumenal chloride in the lysosome is critical for lysosome function. eLife 2017, 6.
- Hipolito, V.E.; Ospina-Escobar, E.; Botelho, R.J. Lysosome remodelling and adaptation during phagocyte activation. Cell. Microbiol. 2018, 20, e12824.
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632.
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296.
- Perera, R.M.; Zoncu, R. The Lysosome as a Regulatory Hub. Annu. Rev. Cell Dev. Biol. 2016, 32, 223–253.
- Ruivo, R.; Anne, C.; Sagné, C.; Gasnier, B. Molecular and cellular basis of lysosomal transmembrane protein dysfunction. Biochim. Biophys. Acta BBA Bioenergy 2009, 1793, 636–649.
- Cox, T.M.; Cachón-González, M.B. The cellular pathology of lysosomal diseases. J. Pathol. 2011, 226, 241–254.
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, N.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658.
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Serkan, E.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433.
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 Senses Lysosomal Amino Acids Through an Inside-Out Mechanism That Requires the Vacuolar H+-ATPase. Science 2011, 334, 678–683.
- Lim, C.-Y.; Zoncu, R. The lysosome as a command-and-control center for cellular metabolism. J. Cell Biol. 2016, 214, 653–664.
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481.
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477.
- Carroll, B.; Dunlop, E.A. The lysosome: A crucial hub for AMPK and mTORC1 signalling. Biochem. J. 2017, 474, 1453–1466.
- Palmieri, M.; Impey, S.; Kang, H.; Di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866.
- Sardiello, M. Transcription factor EB: From master coordinator of lysosomal pathways to candidate therapeutic target in degenerative storage diseases. Ann. N. Y. Acad. Sci. 2016, 1371, 3–14.
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873.
- Sun, X.; Yang, Y.; Zhong, X.Z.; Cao, Q.; Zhu, X.-H.; Zhu, X.; Dong, X.-P. A negative feedback regulation of MTORC1 activity by the lysosomal Ca2+ channel MCOLN1 (mucolipin 1) using a CALM (calmodulin)-dependent mechanism. Autophagy 2018, 14, 38–52.
- Li, R.-J.; Xu, J.; Fu, C.; Zhang, J.; Zheng, Y.G.; Jia, H.; Liu, J.O. Regulation of mTORC1 by lysosomal calcium and calmodulin. eLife 2016, 5, e19360.
- Medina, D.L.; Ballabio, A. Lysosomal calcium regulates autophagy. Autophagy 2015, 11, 970–971.
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Rosato, A.S.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299.
- Blaby-Haas, C.E.; Merchant, S.S. Lysosome-related Organelles as Mediators of Metal Homeostasis. J. Biol. Chem. 2014, 289, 28129–28136.
- Lloyd-Evans, E.; Platt, F.M. Lysosomal Ca2+ homeostasis: Role in pathogenesis of lysosomal storage diseases. Cell Calcium 2011, 50, 200–205.
- Patel, S.; Docampo, R. Acidic calcium stores open for business: Expanding the potential for intracellular Ca2+ signaling. Trends Cell Biol. 2010, 20, 277–286.
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635.
- Lloyd-Evans, E.; Waller-Evans, H. Lysosomal Ca2+Homeostasis and Signaling in Health and Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a035311.
- Lee, H.C.; Aarhus, R. A Derivative of NADP Mobilizes Calcium Stores Insensitive to Inositol Trisphosphate and Cyclic ADP-ribose. J. Biol. Chem. 1995, 270, 2152–2157.
- Churchill, G.C.; Okada, Y.; Thomas, J.M.; Genazzani, A.A.; Patel, S.; Galione, A. NAADP Mobilizes Ca2+ from Reserve Granules, Lysosome-Related Organelles, in Sea Urchin Eggs. Cell 2002, 111, 703–708.
- Christensen, K.A.; Myers, J.T.; Swanson, J.A. pH-dependent regulation of lysosomal calcium in macrophages. J. Cell Sci. 2002, 115, 599–607.
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255.
- Ronco, V.; Potenza, D.M.; Denti, F.; Vullo, S.; Gagliano, G.; Tognolina, M.; Guerra, G.; Pinton, P.; Genazzani, A.A.; Mapelli, L.; et al. A novel Ca2+-mediated cross-talk between endoplasmic reticulum and acidic organelles: Implications for NAADP-dependent Ca2+ signalling. Cell Calcium 2015, 57, 89–100.
- Morgan, A.J.; Platt, F.M.; Lloyd-Evans, E.; Galione, A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 2011, 439, 349–378.
- Narayanaswamy, N.; Chakraborty, K.; Saminathan, A.; Zeichner, E.; Leung, K.; Devany, J.; Krishnan, Y. A pH-correctable, DNA-based fluorescent reporter for organellar calcium. Nat. Methods 2019, 16, 95–102.
- Cao, Q.; Zhong, X.Z.; Zou, Y.; Murrelllagnado, R.D.; Zhu, M.X.; Dong, X.-P. Calcium release through P2X4 activates calmodulin to promote endolysosomal membrane fusion. J. Cell Biol. 2015, 209, 879–894.
- Czibener, C.; Sherer, N.M.; Becker, S.M.; Pypaert, M.; Hui, E.; Chapman, E.R.; Mothes, W.; Andrews, N.W. Ca2+ and synaptotagmin VII–dependent delivery of lysosomal membrane to nascent phagosomes. J. Cell Biol. 2006, 174, 997–1007.
- Pryor, P.R.; Mullock, B.M.; Bright, N.A.; Gray, S.R.; Luzio, J.P. The Role of Intraorganellar Ca2+In Late Endosome–Lysosome Heterotypic Fusion and in the Reformation of Lysosomes from Hybrid Organelles. J. Cell Biol. 2000, 149, 1053–1062.
- Vergarajauregui, S.; Martina, J.A.; Puertollano, R. Identification of the Penta-EF-hand Protein ALG-2 as a Ca2+-dependent Interactor of Mucolipin-1. J. Biol. Chem. 2009, 284, 36357–36366.
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42.
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.-I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884.
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889.
- Sarnat, H.B.; Roth, S.I.; Jimenez, J.F. Neonatal Myotubular Myopathy: Neuropathy and Failure of Postnatal Maturation of Fetal Muscle. Can. J. Neurol. Sci. 1981, 8, 313–320.
- Sawchak, J.A.; Sher, J.H.; Norman, M.G.; Kula, R.W.; Shafiq, S.A. Centronuclear myopathy heterogeneity: Distinction of clinical types by myosin isoform patterns. Neurology 1991, 41, 135.
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830.
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy Is Required to Maintain Muscle Mass. Cell Metab. 2009, 10, 507–515.
- Raben, N.; Hill, V.; Shea, L.; Takikita, S.; Baum, R.; Mizushima, N.; Ralston, E.; Plotz, P. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum. Mol. Genet. 2008, 17, 3897–3908.
- De Duve, C.; Wattiaux, R. Functions of Lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492.
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473.
- Li, W.-W.; Li, J.; Bao, J.-K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136.
- Neff, N.T.; Bourret, L.; Miao, P.; Dice, J.F. Degradation of proteins microinjected into IMR-90 human diploid fibroblasts. J. Cell Biol. 1981, 91, 184–194.
- Cuervo, A.M.; Dice, J.F. Age-related Decline in Chaperone-mediated Autophagy. J. Biol. Chem. 2000, 275, 31505–31513.
- Arstila, A.U.; Trump, B.F. Studies on cellular autophagocytosis. The formation of autophagic vacuoles in the liver after glucagon administration. Am. J. Pathol. 1968, 53, 687–733.
- Mizushima, N.; Ohsumi, Y.; Yoshimori, T. Autophagosome Formation in Mammalian Cells. Cell Struct. Funct. 2002, 27, 421–429.
- Dunn, W.A. Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol. 1994, 4, 139–143.
- Nakatogawa, H. Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol. 2020, 21, 439–458.
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576.
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and Function of Autophagy during Cell Survival and Cell Death. Cold Spring Harb. Perspect. Biol. 2012, 4, a008813.
- Wesselborg, S.; Stork, B. Autophagy signal transduction by ATG proteins: From hierarchies to networks. Cell. Mol. Life Sci. 2015, 72, 4721–4757.
- Aits, S.; Jäättelä, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126, 1905–1912.
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk. Nat. Cell Biol. 2011, 13, 132–141.
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914.
- Wang, L.; Ye, X.; Zhao, T. The physiological roles of autophagy in the mammalian life cycle. Biol. Rev. Camb. Philos. Soc. 2019, 94, 503–516.
- Grimm, C.; Butz, E.; Chen, C.-C.; Wahl-Schott, C.; Biel, M. From mucolipidosis type IV to Ebola: TRPML and two-pore channels at the crossroads of endo-lysosomal trafficking and disease. Cell Calcium 2017, 67, 148–155.
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730.
- Bah, A.; Vergne, I. Macrophage Autophagy and Bacterial Infections. Front. Immunol. 2017, 8, 1483.
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466.
- Fulda, S.; Kogel, D. Cell death by autophagy: Emerging molecular mechanisms and implications for cancer therapy. Oncogene 2015, 34, 5105–5113.
- La Rovere, R.M.; Roest, G.; Bultynck, G.; Parys, J.B. Intracellular Ca 2+ signaling and Ca 2+ microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium 2016, 60, 74–87.
- Bootman, M.D.; Chehab, T.; Bultynck, G.; Parys, J.B.; Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium 2018, 70, 32–46.
- Hu, Y.-X.; Han, X.-S.; Jing, Q. Ca(2+) Ion and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 151–166.
- Kondratskyi, A.; Yassine, M.; Kondratska, K.; Skryma, R.; Slomianny, C.; Prevarskaya, N. Calcium-permeable ion channels in control of autophagy and cancer. Front. Physiol. 2013, 4, 272.
- Vergarajauregui, S.; Connelly, P.S.; Daniels, M.P.; Puertollano, R. Autophagic dysfunction in mucolipidosis type IV patients. Hum. Mol. Genet. 2008, 17, 2723–2737.
- Curcio-Morelli, C.; Charles, F.A.; Micsenyi, M.C.; Cao, Y.; Venugopal, B.; Browning, M.F.; Dobrenis, K.; Cotman, S.L.; Walkley, S.U.; Slaugenhaupt, S.A. Macroautophagy is defective in mucolipin-1-deficient mouse neurons. Neurobiol. Dis. 2010, 40, 370–377.
- Huang, P.; Xu, M.; Wu, Y.; Syeda, A.K.R.; Dong, X.-P.; Mengnan, X.; Yi, W. Multiple facets of TRPML1 in autophagy. Cell Calcium 2020, 88, 102196.
- Scotto Rosato, A.; Montefusco, S.; Soldati, C.; Di Paola, S.; Capuozzo, A.; Monfregola, J.; Polishchuk, E.; Amabile, A.; Grimm, C.; Lombardo, A.; et al. TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKbeta/VPS34 pathway. Nat. Commun. 2019, 10, 5630.
- Li, X.; Rydzewski, N.; Hider, A.; Zhang, X.; Yang, J.; Wang, W.; Gao, Q.; Cheng, X.; Xu, H. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat. Cell Biol. 2016, 18, 404–417.
- Sun, T.; Wang, X.; Lu, Q.; Ren, H.; Zhang, H. CUP-5, The C. elegans ortholog of the mammalian lysosomal channel protein MLN1/TRPML1, is required for proteolytic degradation in autolysosomes. Autophagy 2011, 7, 1308–1315.
- Yang, Y.; Xu, M.; Zhu, X.; Yao, J.; Shen, B.; Dong, X.-P. Lysosomal Ca2+ release channel TRPML1 regulates lysosome size by promoting mTORC1 activity. Eur. J. Cell Biol. 2019, 98, 116–123.
- Wang, W.; Gao, Q.; Yang, M.; Zhang, X.; Yu, L.; Lawas, M.; Li, X.; Bryant-Genevier, M.; Southall, N.T.; Marugan, J.; et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc. Natl. Acad. Sci. USA 2015, 112, E1373–E1381.
- Kim, H.J.; Soyombo, A.A.; Tjon-Kon-Sang, S.; So, I.; Muallem, S. The Ca2+Channel TRPML3 Regulates Membrane Trafficking and Autophagy. Traffic 2009, 10, 1157–1167.
- Kim, S.W.; Kim, D.H.; Park, K.S.; Kim, M.K.; Park, Y.M.; Muallem, S.; So, I.; Kim, H.J. Palmitoylation controls trafficking of the intracellular Ca2+ channel MCOLN3/TRPML3 to regulate autophagy. Autophagy 2019, 15, 327–340.
- Choi, S.; Kim, H.J. The Ca2+ channel TRPML3 specifically interacts with the mammalian ATG8 homologue GATE16 to regulate autophagy. Biochem. Biophys. Res. Commun. 2014, 443, 56–61.
- Pereira, G.J.S.; Hirata, H.; Fimia, G.M.; Carmo, L.G.D.; Bincoletto, C.; Han, S.W.; Stilhano, R.S.; Ureshino, R.P.; Bloor-Young, D.; Churchill, G.; et al. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Regulates Autophagy in Cultured Astrocytes. J. Biol. Chem. 2011, 286, 27875–27881.
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107.
- Gómez-Suaga, P.; Luzón-Toro, B.; Churamani, D.; Zhang, L.; Bloor-Young, D.; Patel, S.; Woodman, P.G.; Churchill, G.C.; Hilfiker, S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 2011, 21, 511–525.
- Lin, P.-H.; Duann, P.; Komazaki, S.; Park, K.H.; Li, H.; Sun, M.; Sermersheim, M.; Gumpper, K.; Parrington, J.; Galione, A.; et al. Lysosomal Two-pore Channel Subtype 2 (TPC2) Regulates Skeletal Muscle Autophagic Signaling. J. Biol. Chem. 2015, 290, 3377–3389.
- Cang, C.; Zhou, Y.; Navarro, B.; Seo, Y.-J.; Aranda, K.; Shi, L.; Battaglia-Hsu, S.; Nissim, I.; Clapham, D.E.; Ren, D. mTOR Regulates Lysosomal ATP-Sensitive Two-Pore Na+ Channels to Adapt to Metabolic State. Cell 2013, 152, 778–790.
- Tian, X.; Gala, U.; Zhang, Y.; Shang, W.; Jaiswal, S.N.; Di Ronza, A.; Jaiswal, M.; Yamamoto, S.; Sandoval, H.; DuRaine, L.; et al. A voltage-gated calcium channel regulates lysosomal fusion with endosomes and autophagosomes and is required for neuronal homeostasis. PLoS Biol. 2015, 13, e1002103.
- Gkika, D.; Prevarskaya, N. TRP channels in prostate cancer: The good, the bad and the ugly? Asian J. Androl. 2011, 13, 673–676.
- Karacosta, L.G.; Foster, B.A.; Azabdaftari, G.; Feliciano, D.M.; Edelman, A.M. A Regulatory Feedback Loop Between Ca2+/Calmodulin-dependent Protein Kinase Kinase 2 (CaMKK2) and the Androgen Receptor in Prostate Cancer Progression. J. Biol. Chem. 2012, 287, 24832–24843.
- Liu, L.H.; Boivin, G.P.; Prasad, V.; Periasamy, M.; Shull, G.E. Squamous Cell Tumors in Mice Heterozygous for a Null Allele of Atp2a2, Encoding the Sarco(endo)plasmic Reticulum Ca2+-ATPase Isoform 2 Ca2+Pump. J. Biol. Chem. 2001, 276, 26737–26740.
- Monteith, G.R.; Davis, F.M.; Roberts-Thomson, S.J. Calcium Channels and Pumps in Cancer: Changes and Consequences. J. Biol. Chem. 2012, 287, 31666–31673.
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375.
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471.
- Galluzzi, L.; Pietrocola, F.; Pedro, J.M.B.-S.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880.
- Santana-Codina, N.; Mancias, J.D.; Kimmelman, A.C. The Role of Autophagy in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 19–39.
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 1–16.
- Levy, J.M.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020, 27, 843–857.
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734.
- Towers, C.G.; Thorburn, A. Targeting the Lysosome for Cancer Therapy. Cancer Discov. 2017, 7, 1218–1220.
- Geisslinger, F.; Müller, M.; Vollmar, A.M.; Bartel, K. Targeting Lysosomes in Cancer as Promising Strategy to Overcome Chemoresistance—A Mini Review. Front. Oncol. 2020, 10, 1156.
- Faris, P.; Shekha, M.; Montagna, D.; Guerra, G.; Moccia, F. Endolysosomal Ca(2+) Signalling and Cancer Hallmarks: Two-Pore Channels on the Move, TRPML1 Lags Behind! Cancers 2018, 11, 27.
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2017, 7, 3–17.
- Jung, J.; Cho, K.; Naji, A.K.; Clemons, K.N.; Wong, C.O.; Villanueva, M.; Gregory, S.; Karagas, N.E.; Tan, L.; Liang, H.; et al. HRAS-driven cancer cells are vulnerable to TRPML1 inhibition. EMBO Rep. 2019, 20, e46685.
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142.
- Jung, J.; Venkatachalam, K. TRPML1 and RAS-driven cancers—Exploring a link with great therapeutic potential. Channels 2019, 13, 374–381.
- Elliott, I.A.; Dann, A.M.; Xu, S.; Kim, S.S.; Abt, E.R.; Kim, W.; Poddar, S.; Moore, A.; Zhou, L.; Williams, J.L.; et al. Lysosome inhibition sensitizes pancreatic cancer to replication stress by aspartate depletion. Proc. Natl. Acad. Sci. USA 2019, 116, 6842–6847.
- Xu, M.; Almasi, S.; Yang, Y.; Yan, C.; Sterea, A.M.; Syeda, A.K.R.; Shen, B.; Derek, C.R.; Huang, P.; Gujar, S.; et al. The lysosomal TRPML1 channel regulates triple negative breast cancer development by promoting mTORC1 and purinergic signaling pathways. Cell Calcium 2019, 79, 80–88.
- Kasitinon, S.Y.; Eskiocak, U.; Martin, M.; Bezwada, D.; Khivansara, V.; Tasdogan, A.; Zhao, Z.; Mathews, T.; Aurora, A.B.; Morrison, S.J. TRPML1 Promotes Protein Homeostasis in Melanoma Cells by Negatively Regulating MAPK and mTORC1 Signaling. Cell Rep. 2019, 28, 2293–2305.e9.
- Santoni, G.; Maggi, F.; Morelli, M.B.; Santoni, M.; Marinelli, O. Transient Receptor Potential Cation Channels in Cancer Therapy. Med. Sci. 2019, 7, 108.
- Morelli, M.B.; Nabissi, M.; Amantini, C.; Tomassoni, D.; Rossi, F.; Cardinali, C.; Santoni, M.; Arcella, A.; Oliva, M.A.; Santoni, A.; et al. Overexpression of transient receptor potential mucolipin-2 ion channels in gliomas: Role in tumor growth and progression. Oncotarget 2016, 7, 43654–43668.
- Santoni, G.; Santoni, M.; Maggi, F.; Marinelli, O.; Morelli, M.B. Emerging Role of Mucolipins TRPML Channels in Cancer. Front. Oncol. 2020, 10, 659.
- Atakpa, P.; Van Marrewijk, L.M.; Apta-Smith, M.; Chakraborty, S.; Taylor, C.W. GPN does not release lysosomal Ca2+ but evokes Ca2+ release from the ER by increasing the cytosolic pH independently of cathepsin C. J. Cell Sci. 2019, 132, jcs223883.