Polyglutamine spinocerebellar ataxias (PolyQ SCAs) are a group of 6 rare autosomal dominant diseases, which arise from an abnormal CAG repeat expansion in the coding region of their causative gene. These neurodegenerative ataxic disorders are characterized by progressive cerebellar degeneration, which translates into progressive ataxia, the main clinical feature, often accompanied by oculomotor deficits and dysarthria. Currently, PolyQ SCAs treatment is limited only to symptomatic mitigation, and no therapy is available to stop or delay the disease progression, which culminates with death.
- polyglutamine disorders
- spinocerebellar ataxia
- gene therapy
- gene augmentation
- gene silencing
- gene editing
1. Introduction
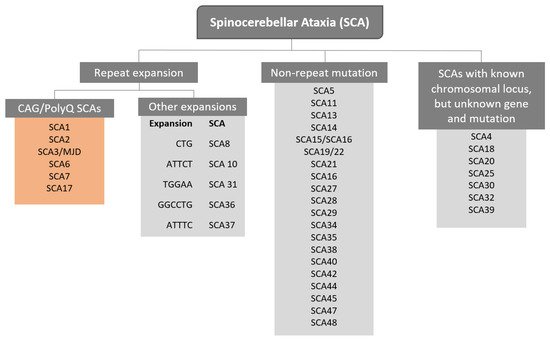
The term Spinocerebellar refers to the spinal cord and cerebellum, and ataxia means “absence of coordination”. So, Spinocerebellar Ataxias (SCAs) are a group of degenerative diseases of the nervous system in which progressive ataxia occurs [1]. Until now, there are more than 40 genetically different SCA subtypes identified, which include inherited autosomal recessive cerebellar ataxias, autosomal dominant spinocerebellar ataxias, and X-linked ataxias [2,3,4]. The nomenclature is given by the term SCA followed by a number, representative of the chronological order of the causative gene or disease locus discovery. The latest discovered subtype is SCA 48 [5], however, some numbers are vacant, as some subtypes overlap or share the same muted gene e.g., SCA15 and SCA16, and SCA19 and SCA22 [6]. SCAs are categorized into two subgroups according to the causative genetic origin: the non-repeat mutations and repeat expansions SCAs (Figure 1). SCAs are rare inherited diseases affecting approximately 1–5:100,000 persons worldwide [7]. The most common is SCA3/MJD followed by SCA2 and SCA6, respectively [8]. The three countries where SCAs are more prevalent are: Portugal with the highest rates in population registering 5.6:100,000 [8], followed by Norway registering rates of 4.2:100,000 [9], and Japan registering rates of 5:100,000 [10]. In this review, we are going to focus specifically on the autosomal dominant SCAs with CAG expansion, also known as Polyglutamine (PolyQ) SCAs.
PolyQ SCAs are caused by an abnormal CAG trinucleotide repeat expansion within the coding region of the causative gene that encodes for the glutamine amino acids, generating an expanded polyglutamine protein. The repeat expansion size varies from individuals, being related to the disease severity, and the age onset, that is, individuals with higher CAG repetitions display an early onset of the symptoms, which are also more severe [11]. PolyQ SCAs are also characterized by the anticipation of the age onset, explained by intergenerational instability biased towards expansions. Additionally, somatic mosaicism was also reported as being associated with the age of onset and the severity of the symptoms [12]. The disease onset is usually in the fourth decade of life, with a life expectancy that can vary between 10–15 years [13]. The first symptoms are usually gait ataxia, frequently followed by limb incoordination, speech disturbance, and oculomotor abnormalities [14] according to the SCA subtype. Although the symptomatology is similar between PolyQ SCAs, different repeat size and genes are responsible for the SCA subtype (Table 1).
Table 1. Classification of autosomal dominant spinocerebellar ataxia with CAG expansions.
|
PolyQ |
Gene |
Locus |
Protein |
Molecular Function |
Repeats |
||
|---|---|---|---|---|---|---|---|
|
Normal |
Intermediate |
Disease |
|||||
|
SCA1 |
ATXN1 |
6p22.3 |
Ataxin-1 |
Transcription Factor Interactor |
9–39 |
40 |
41–83 |
|
SCA2 |
ATXN2 |
12q24.12 |
Ataxin-2 |
RNA metabolism |
<31 |
31–33 |
34–200 |
|
SCA3/MJD |
ATXN3 |
14q32.12 |
Ataxin-3 |
Deubiquitinase |
12–44 |
45–55 |
56–86 |
|
SCA6 |
CACNA1A |
19p13.13 |
Calcium voltage-gated channel subunit alpha 1 G |
Channel and Transcription Factor |
<18 |
19 |
20–33 |
|
SCA7 |
ATXN7 |
3p14.1 |
Ataxin-7 |
Transcription Factor (SAGA Complex) |
4–19 |
28–33 |
34–460 |
|
SCA17 |
TBP |
6q27 |
TATA box-binding protein |
Transcription Factor |
25–40 |
- |
41–66 |
SCA–Spinocerebellar ataxia; MJD–Machado-Joseph Disease; ATXN–Ataxin gene; CACNA1A–Calcium voltage-gated channel subunit α1 G; TBP–TATA box-binding protein; p–chromosome shorter arm; q–chromosome longer arm; SAGA complex–SPT-ADA-GCN5 acetyltransferase [1,2,3].
2. Gene Therapy Augmentation Strategies
2.1. Strategies Activating Autophagy
2.2. Neuroprotective Strategies
| Disease | Molecular Target | Gene Delivery System | Strategy | References |
|---|---|---|---|---|
| SCA1 | Homer-3 | AAV vectors | Autophagy | [45] |
| Ataxin-1 like | AAV vectors | Neuroprotection | [47] | |
| SCA3 | CYP46A1 | AAV vectors | Autophagy | [42] |
| Beclin-1 | Lentiviral vector | Autophagy | [44] | |
| Calpastatin | AAV vectors | Proteolytic cleavage | [51] | |
| Wild-type ataxin-3 | Lentiviral vector | Neuroprotection | [48] | |
| Ataxin-2 | Lentiviral vector | Neuroprotection | [49] | |
| CRAG | Lentiviral vector | Neuroprotection | [52] | |
| NPY | AAV vectors | Neuroprotection | [53] |
3. Gene Silencing Strategies
PolyQ SCAs have their underlying cause in single genetic factors, which influences a diverse set of downstream molecular pathways that contribute for disease progression [16]. Additionally, in vivo studies, regarding mutant ATXN3 RNA-derived toxicity, have reported that the expression of untranslated transcripts with abnormally expanded CAG repeats lead to cell degeneration in Drosophila, Caenorhabditis elegans and mouse models [54,55,56]. This toxicity may be due to (i) the formation of expanded CAG RNAs foci, which can sequester proteins implicated in alternative splicing [57]; (ii) interference with nucleolar function [58] or (iii) silencing of the expression of certain genes [59]. The length of untranslated CAG transcripts was also shown to directly influence the toxicity of such RNA molecules, as the expression of transcripts with increased CAG length, deteriorated motor phenotype [55]. Considering these findings, silencing the expression of such pathological RNAs will result in a lack of toxic protein translation, while also eliminating the toxicity exerted by the RNA molecules themself. Therefore, the most straightforward therapeutic approach for PolyQ SCAs would be to silence the expression of the gene containing the disease-causing mutation. This may be achieved by promoting RNA degradation, skipping the mutant exon, impairing protein translation, correcting the pathological mutation or preventing gene translation altogether. Ultimately, such gene silencing strategies would allow to act at the earliest steps possible, admittedly preventing disease onset or progression [16].
3.1. RNAi-Based Gene Silencing Strategies
3.1.1. Short Hairpin and Small Interfering RNAs Mediated Silencing
| Disease | Target | Allele Specificity | Technology | Experimental Systems | Delivery | References |
|---|---|---|---|---|---|---|
| SCA1 | Ataxin-1 | Non-specific | shRNA | SCA1 transgenic mouse model | AAV-mediated transduction | [66] |
| SCA3/MJD | Mutant Ataxin-3 |
Allele-specific | siRNA | HEK 293T cells | Transfection | [73] |
| Mutant Ataxin-3 | Allele-specific | shRNA | LV-induced SCA3/MJD rat model | LV-mediated transduction | [72] | |
| Mutant Ataxin-3 | Allele-specific | siRNA | SCA3/MJD transgenic mouse model | SNALPs-mediated transduction | [76] | |
| Mutant Ataxin-3 | Allele-specific | shRNA | Patient derived fibroblasts | LV-mediated transduction | [79] | |
| Ataxin-3 | Non-specific | shRNA | LV-induced SCA3/MJD rat model | LV-mediated transduction | [48] | |
| SCA7 | Mutant Ataxin-7 | Allele-specific | siRNA | Patient derived fibroblasts | Transfection | [77] |
| Mutant Ataxin-7 | Allele-specific | siRNA | Patient derived fibroblasts | Transfection | [78] | |
| Mutant Ataxin-7 | Allele-specific | shRNA | Patient derived fibroblasts | LV-mediated transduction | [79] |
3.1.2. MicroRNA-Mediated Silencing
| Disease | microRNAs | Target | Experimental System | Delivery | References |
|---|---|---|---|---|---|
| SCA1 | Artificial miRNA | ATXN1 | C2C12 cells, SCA1 transgenic mouse model and non-human primates | AVV-mediated transduction | [67,68,81,83] |
| miR-19, miR-101 and miR-130 mimic | ATXN1 | HEK 293T, HeLa and MCF7 cells | Transfection | [91] | |
| miR-144 mimic | ATXN1 | HEK 293T cells | Transfection | [92] | |
| SCA3/MJD | Artificial miRNA | ATXN3 | SCA3/MJD transgenic mouse model | AAV-mediated transduction | [82,86] |
| miR-25 mimic | ATXN3 | HEK 293T and SH-S5Y5 cells | Transfection | [87] | |
| miR-9, miR-181a and miR-494 mimics | ATXN3 | HEK 293T cells and LV-induced SCA3/MJD mouse model | LV-mediated transduction | [88] | |
| SCA6 | miR-3191-5p | CACNA1A | AAV-induced SCA6 mouse model | AAV-mediated transduction | [89] |
| SCA7 | Artificial miRNA | ATXN7 | SCA7 transgenic mouse model | AAV-mediated transduction | [84,85] |
| miR-124 mimic | Lnc-SCA7 and ataxin-7 | N2a cells | Transfection | [90] |
3.2. Antisense Oligonucleotides
Antisense oligonucleotides (ASOs) are synthetic single-stranded DNA molecules, capable of hybridizing to target mRNA molecules through Watson-Crick base pairing and alter their functions, and thus allowing to artificially modulate gene expression via different mechanisms [93]. These mechanisms may be divided in two groups: RNAse H-dependent mRNA degradation and RNAse H-independent. In order to mediate RNAse H-dependent mRNA degradation, a portion of nucleotides in the 2′ position of the ASOs molecules must remain unmodified [94,95]. Following the formation of the mRNA:DNA(ASO) duplex and its recognition by RNAse H, the target mRNA molecule is cleaved, while the artificial oligonucleotide molecule remains intact [95]. In the case of RNAse H-independent RNA modulation, completely 2′-modified ASOs may be used to mediate several processes where mRNA degradation is not the outcome. This mechanism can be employed to modulate mRNA splicing events, which in the context of PolyQ SCAs could enable skipping the exons containing the pathological mutation [96,97].
ASOs have been employed in several PolyQ SCAs studies as a potential silencing strategy (Table 6). In 2013, Evers and colleagues demonstrated that ASOs were able to mediate exon skipping of the ATXN3 exon containing the CAG repeats in cell and mouse models, without any apparent deleterious effects [96]. Additionally, the same group also showed that this exon skipping strategy in a transgenic SCA3/MJD mouse model led to a reduction in ataxin-3 insolubility and nuclear accumulation of the protein [98]. A study employing ASOs to promote ataxin-3 RNA degradation resulted in a reduction of mutant ataxin-3 protein levels in cell and mouse models [99]. Following this previous study, the most promising ASO was chosen and delivered to a SCA3/MJD mouse model. Upon ASO delivery, mitigation of several disease-associated phenotypes was observed [100].
Regarding other PolyQ SCAs, recent studies applying ASOs mediated RNA suppression to reduce target gene expression and ameliorate disease phenotypes in mouse models have yielded promising results. In fact, studies in SCA1 and SCA2 mouse models, revealed that upon intracerebroventricular (IVC) injection, expression of ATXN1 and ATXN2 mRNA levels were significantly decrease and motor deficits mitigated [101,102]. Noteworthy, ASO-mediated ATXN7 knockdown, in the eye of SCA7 mice, resulted in a considerable reduction of Ataxin-7 expression and protein aggregation, as well as the amelioration of several visual impairments [103].
More recently, a study performed in SCA3/MJD and SCA1 mouse models employed an ASO molecule, entitled (CUG)7, which had been previously developed to target the CAG stretches in Huntingtin [104]. In this study, researchers were able to successfully reduce the protein levels of mutant Ataxin-3 in SCA3/MJD patient derived fibroblasts and mouse model. As for SCA1, (CUG)7, was also able to reduce the protein levels on mutant ataxin-1 in both cell and mouse models [105].
In recent years ASOs-based approaches have seen a rise in interest. Promising results in several PolyQ SCAs established ASOs as a viable alternative to RNAi-based strategies. Notably, ASOs have greatly expanded their range in target diseases and improved upon existing ones in the last few years. Exon skipping mediated by ASOs may also become instrumental in the future potentially resolving mutant RNA derived toxicity, which has been identified as a possible pathological mechanism in PolyQ SCAs. Finally, multi-PolyQ disease approaches using ASOs such as the one developed by Kourkouta and colleagues [105] may mark the rise of ASOs as the future gold standard in oligonucleotide-mediated gene silencing.
| Disease | Target | Mechanism | Delivery Method | References |
|---|---|---|---|---|
| SCA1 | Ataxin-1 | RNAse-H dependent degradation | ICV | [101] |
| Ataxin-1 | Translation hindrance | ICV | [105] | |
| SCA2 | Ataxin-2 | RNAse-H dependent degradation | ICV | [102] |
| SCA3/MJD | Ataxin-3 | Exon 9 and 10-skipping | ICV | [96] |
| Ataxin-3 | RNAse-H dependent degradation | ICV | [99] | |
| Ataxin-3 | Exon10-skiping | ICV | [98] | |
| Ataxin-3 | Translation hindrance and Exon10-skiping | ICV | [105] | |
| SCA7 | Ataxin-7 | RNAse-H dependent degradation | IVI | [103] |
This entry is adapted from the peer-reviewed paper 10.3390/ijms22084249