Iron is essential for energy metabolism, and states of iron deficiency or excess are detrimental for organisms and cells. Therefore, iron and carbohydrate metabolism are tightly regulated. Serum iron and glucose levels are subjected to hormonal regulation by hepcidin and insulin, respectively. Hepcidin is a liver-derived peptide hormone that inactivates the iron exporter ferroportin in target cells, thereby limiting iron efflux to the bloodstream. Insulin is a protein hormone secreted from pancreatic β-cells that stimulates glucose uptake and metabolism via insulin receptor signaling. There is increasing evidence that systemic, but also cellular iron and glucose metabolic pathways are interconnected.
- hepcidin
- ferroportin
- insulin
- adipokines
- IRP1
- IRP2
1. Iron and Energy Metabolism
Iron is a transition metal with critical biological functions [1]. In mammals, most of body iron is present in hemoglobin of red blood cells and mediates oxygen transport. Significant amounts of iron are also present within myoglobin of skeletal muscle cells. Other cell types require smaller quantities of iron for utilization by several metalloproteins. These include metabolic enzymes and oxidoreductases, which catalyze electron transfer reactions. The activity of mitochondrial aconitase, an enzyme catalyzing conversion of citrate to isocitrate in the tricarboxylic acid (TCA) cycle, depends on a 4Fe-4S cluster in its active site. Moreover, four out of five complexes in the mitochondrial electron transport chain contain hemoproteins (such as cytochromes) or iron–sulfur cluster proteins. Thus, iron is essential for cellular energy metabolism.
Cell culture experiments showed that iron depletion inhibits not only mitochondrial aconitase, but also other enzymes of the TCA cycle such as citrate synthase, isocitrate dehydrogenase and succinate dehydrogenase [2]. This decreases formation of NADH and ATP, and also reduces oxygen consumption in the electron transport chain. To compensate for the inhibition in respiration, the iron-depleted cell increases glycolysis for ATP synthesis. On the other hand, excessive iron negatively affects mitochondrial function. Thus, dietary iron overload of mice decreases oxidative phosphorylation in liver mitochondria and also promotes mitochondrial disfunction due to oxidative stress [3]. This is consistent with the notion that while iron is an essential nutrient, it may also become a potent biohazard by promoting oxidative stress [4]. The Janus face of iron indicates that balanced iron metabolism is imperative for health [5]. Mechanisms underlying regulation of systemic and cellular iron metabolism are summarized below.
2. Overview of Glucose Metabolism
Glucose is the principal source for metabolic energy. It is mainly acquired from the diet as breakdown product of food macromolecules but can also be mobilized from glycogen stores or synthesized from other metabolites. Ingested glucose is absorbed by intestinal enterocytes via SGLT1 (sodium–glucose co-transporter 1) [6] and is released to plasma via GLUT2, a member of the GLUT family of facilitative glucose transporters [7]. GLUT transporters also account for cellular uptake of circulating glucose by a passive diffusion mechanism, which is driven by the lower intracellular glucose concentration. Skeletal muscle cells, cardiomyocytes and adipocytes acquire glucose via the insulin-regulated glucose transporter GLUT4, while hepatocytes primarily utilize GLUT2 [8]. Pancreatic β cells take up glucose mostly via GLUT2 in rodents and GLUT1 and GLUT3 in humans [9].
The fate of intracellular glucose differs among cell types. All cells can metabolize glucose via glycolysis to pyruvate, which is further converted to acetyl-CoA to enter the TCA cycle. This key metabolic pathway is prominent in energy-demanding muscle cells. Adipocytes utilize acetyl-CoA for fatty acid biosynthesis to store energy. Hepatocytes mainly convert glucose to glycogen for energy storage and can also synthesize glucose by gluconeogenesis. Glucose uptake by pancreatic β cells is critical for insulin synthesis and systemic glucose regulation.
Plasma glucose levels are maintained within a narrow range by the pancreatic hormones glucagon and insulin. Hypoglycemia triggers secretion of glucagon by pancreatic α cells, which promotes glycogenolysis and gluconeogenesis in the liver, and lipolysis in adipose tissue; these responses aim to restore euglycemia. On the other hand, hyperglycemia triggers secretion of insulin from pancreatic β cells, which promotes glucose uptake for energy production and anabolic processes such as glycogen synthesis and lipogenesis in the liver, muscles and adipose tissue (Figure 1).
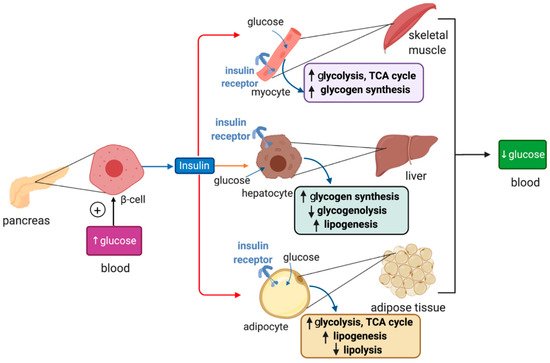
Figure 1. Hormonal regulation of glucose metabolism by insulin. Insulin is synthesized in pancreatic β cells in response to hyperglycemia. It binds to insulin receptors in target cells (red arrows) such as skeletal muscle cells, hepatocytes and adipocytes and induces signaling pathways that promote glucose uptake, catabolism or storage. This reduces plasma glucose levels.
Insulin binds to insulin receptors and activates PI3K/Akt signaling cascades [10]. In skeletal muscle cells and adipocytes insulin, signaling promotes translocation of GLUT4-containing storage vesicles to the plasma membrane for glucose absorption. Moreover, in skeletal muscle cells, insulin signaling inactivates the inhibitory GSK3 (glycogen synthase kinase 3), which restores glycogen synthase activity and allows glycogen synthesis. In adipocytes, insulin signaling inhibits HSL (hormone-sensitive lipase) to suppress lipolysis. In hepatocytes, insulin signaling targets GSK3 to induce glycogen synthesis and phosphorylase kinase to inhibit glycogenolysis. In addition, it activates protein synthesis, inhibits gluconeogenesis and stimulates lipogenesis.
3. Iron and Glucose Metabolism in Human Disease
Iron overload is an established risk factor for diabetes [11][12]. This is vividly illustrated in the high (20–50%) frequency of diabetes in patients with iron overload disorders such as hereditary hemochromatosis [13] or β-thalassemia [14], which is related to both insulin resistance and destruction of pancreatic β-cells. Moreover, epidemiological studies provided links between aberrant iron metabolism and the metabolic syndrome (MetS), a pathologic condition defined by the combined manifestation of at least three of the following conditions: abdominal obesity, hyperglycemia due to insulin resistance, hyperlipidemia and hypertension. Thus, many MetS patients develop mild systemic iron overload characterized by excess liver iron, the presence of serum non-transferrin bound iron and hyperferritinemia [15][16][17][18]. The combination of unexplained iron overload with insulin resistance is also referred to as dysmetabolic iron overload syndrome (DIOS) and has a prevalence of 15–30% among MetS patients [19]. On the other hand, obesity is also considered as a risk factor for iron deficiency, and some obese patients develop anemia; most likely, these responses are associated with inflammatory induction of hepcidin [20].
Non-alcoholic fatty liver disease (NAFLD) represents the hepatic component of the MetS and constitutes the most frequent liver disease in Western countries [21][22]. It is characterized by excessive fat deposition in hepatocytes (steatosis) in the absence of other causes of liver injury (i.e., alcohol abuse or viral hepatitis). In many NAFLD patients, liver disease progresses from simple steatosis to non-alcoholic steatohepatitis (NASH), a chronic inflammatory condition that may further lead to liver fibrosis, cirrhosis and hepatocellular carcinoma (HCC). Excess hepatic iron is a risk factor for progression of NAFLD to NASH, liver cirrhosis and HCC [23]. Thus, liver iron deposition is more frequent in individuals with NASH-related cirrhosis with HCC than in HCC-free controls [24]. Consequently, manipulation of iron metabolic pathways has been proposed to offer a promising therapeutic target [25].
In some occasions, interventions leading to reduction of iron stores (phlebotomy, treatment with iron-chelating drugs or use of iron-deficient diets) have improved insulin sensitivity in MetS patients [26][27] and rodent models [28][29]. However, clinical data on iron depletion strategies to improve insulin sensitivity or ameliorate metabolic liver disease have had mixed outcomes and are largely inconclusive [11][30]. Therefore, a better understanding of the complex mechanisms linking iron and glucose metabolism at the systemic and cellular levels is required to develop and improve iron-related therapeutic interventions.
4. Links Between Systemic Iron and Glucose Metabolism
Levels of both plasma iron and glucose are subjected to negative hormonal regulation by hepcidin and insulin, respectively [31]. This analogy indicates that iron and glucose metabolism may be interconnected. In fact, there are several examples supporting this view. Thus, insulin directly induces hepcidin expression in hepatocytes via STAT3; this response is attenuated and may contribute to iron overload in a rat model of type 2 diabetes induced by combination of streptozotocin treatment and high-fat diet [32]. Likewise, glucose intake was shown to augment circulating hepcidin and decrease serum iron levels in healthy volunteers [33]. It is unclear whether this was a result of systemic hepcidin induction in hepatocytes or local hepcidin production by insulin-secreting pancreatic β-cells. In vitro studies showed that glucose stimulates hepcidin expression in rat INS-1E insulinoma but not human HepG2 hepatoma cells [33]. Another study found that high, glucotoxic glucose levels rather suppress hepcidin in islets from db/db mice, which lack the receptor of the insulin-sensitizing hormone leptin, and in mouse Min6 insulinoma cells [34]. This response appears to promote iron overload, oxidative stress, mitochondrial depolarization and β-cell dysfunction [35]. The underlying mechanisms are not well understood; nevertheless, the metabolic phenotypes of obese leptin receptor-deficient db/db [35], leptin-deficient ob/ob [29] and KKAy [36] mice can be improved by dietary iron restriction.
Metabolic cues can also modulate systemic hepcidin expression in the liver. Thus, mice respond to starvation by transcriptional induction of Hamp mRNA, possibly to preserve sufficient iron levels in tissues with high metabolic needs [37]. The mechanism involves the transcription factor CREBH and the transcriptional coactivator PPARGC1A, which are activated by gluconeogenic signals. In another example, leptin was reported to increase HAMP mRNA expression in human Huh7 hepatoma cells via STAT3 [38]. In line with this finding, ob/ob and db/db mice exhibit lower serum hepcidin levels [39][40], while administration of recombinant leptin to ob/ob mice increased hepatic Hamp mRNA and serum hepcidin [39]. It should also be noted that wild-type mice on obesogenic high-fat diets exhibit reduced Hamp mRNA expression and tend to develop mild iron deficiency [41][42]. Hepcidin-independent suppression of iron absorption is another contributing factor [43]. On the other hand, high-fat diets have been shown to promote Hamp mRNA induction and iron overload in rats [44][45]. The upregulation of Hamp mRNA is consistent with the fact that obesity is associated with chronic low grade inflammation and release of hepcidin-inducing inflammatory cytokines such as IL-6 by adipocytes [46][47]. Nevertheless, the reasons for the discrepancies between mouse and rat models are unclear.
Tmprss6-/- mice express high levels of hepcidin due to disruption of the Tmprss6 gene encoding the hepcidin inhibitor matriptase-2 [48]. These animals, which offer a model of iron-refractory iron deficiency anemia, are protected against high-fat diet-induced obesity and liver steatosis [49]. Moreover, they exhibit improved glucose tolerance and insulin sensitivity, enhanced fat lipolysis and reduced adipocyte hypertrophy. This phenotype was reversed upon injection of an anti-HJV antibody blocking hepcidin overexpression and was enhanced by injection of iron dextran that further stimulates hepcidin. Interestingly, iron dextran also improved metabolism in wild-type mice via hepcidin induction [49]. Considering that iron overload is generally associated with metabolic dysfunction (see below), this finding appears paradoxical. Nevertheless, feeding wild-type or hepcidin-deficient Hjv-/- mice with a combination of high-iron and high-fat diet was also protective against obesity, liver steatosis, hyperglycemia and hypercholesterolemia triggered by the high-fat diet alone [41]. These data underline the complexity of metabolic responses to alterations in hepcidin and systemic iron levels. It is conceivable that additional factors can shift the balance towards improved or worsened metabolic function when hepcidin and systemic iron levels fluctuate.
One potential factor is the intestinal microbiota. Under physiological symbiotic conditions, the gut is the source of microbial-derived nutrients and metabolites, such as short-chain fatty acids (SCFA) and secondary bile acids, which cross the intestinal barrier and reach the liver to control metabolic pathways. However, intake of high-fat and low-fiber diets promotes dysbiosis, inflammation and changes in microbiome composition, favoring propagation of pathogenic bacteria. In this setting, alterations in the intestinal barrier allow increased absorption of branched chain amino acids (BCAA) and lipopolysaccharide (LPS) at the expense of SCFA and bile acids. These responses contribute to peripheral insulin resistance, immunological dysfunction and liver steatosis [50][51][52]. Iron is emerging as an important regulator of the microbiome [53]. While intestinal bacteria are known to utilize luminal dietary iron for growth, it appears that they also respond to tissue iron levels. Thus, variations in microbiome composition have been reported in humans [54] and mice [55] receiving parenteral iron, and in mice with genetically altered iron homeostasis [56]. Conversely, intestinal bacteria can control dietary iron absorption and iron homeostasis in the host by secreting metabolites modulating expression of HIF2α in enterocytes [57].
Another factor that may affect the interplay between systemic iron and glucose metabolism is the iron content of adipose tissue, which can modulate synthesis of insulin-regulating adipokines (Figure 2). Thus, adipocyte iron induces expression of the insulin-inhibitory adipokines resistin [58] and RBP-4 (retinol-binding protein 4) [59], causing insulin resistance. Experiments in 3T3-L1 adipocytes and mice showed that iron also inhibits expression of insulin-sensitizing leptin via phosphorylation-dependent inactivation of CREB [60]. In addition, systemic iron overload suppresses adiponectin, another insulin-sensitizing adipokine, by a mechanism involving the FOXO-1 (forkhead box protein O1) transcription factor [61]. However, Hfe-/- mice, a model of hereditary hemochromatosis, exhibited increased adiponectin expression and improved glucose tolerance. This was attributed to reduced iron content of adipose tissue, in spite of systemic iron overload [61]. Mechanistically, hepcidin deficiency would allow unrestricted ferroportin expression and iron efflux from adipocytes. Nevertheless, further experiments with adipocyte-specific ferroportin knockout mice were not supportive to this view [62], emphasizing the need for more studies to clarify the disparity. This could be related to genetic background differences in mouse strains, which is known to influence iron content and function of adipose tissue [63].
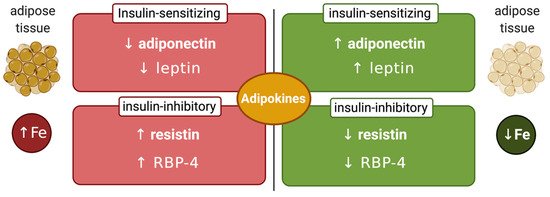
Figure 2. Iron-dependent regulation of adipokines. Increased adipose tissue iron levels inhibit insulin-sensitizing adiponectin and leptin and stimulate insulin-inhibitory resistin and retinol-binding protein 4 (RBP-4). Conversely, reduced adipose tissue iron levels stimulate insulin-sensitizing adiponectin and leptin and inhibit insulin-inhibitory resistin and RBP-4.
Adipose tissue iron may be affected by additional signals. One of them is locally produced adipose tissue hepcidin, which has been documented in obese humans [64] and mice [65]. Additionally, there is evidence that a subset of M2-like anti-inflammatory resident macrophages with high iron storage capacity is present in the healthy adipose tissue and serves to spare adipocytes from iron overload. However, in obesity, this subpopulation acquires M1-like pro-inflammatory features and loses its iron storage capacity, favoring adipocyte iron overload [66]. This important topic is extensively discussed in a comprehensive review article [67].
5. Links between Cellular Iron and Glucose Metabolism
At the cellular level, insulin was reported to stimulate TfR1-mediated iron uptake in primary rat adipocytes [68] and in human HepG2 hepatoma cells [69]. Importantly, excessive cellular iron accumulation has been associated with insulin resistance in primary rat adipocytes [70], primary mouse hepatocytes [71] or rat H9c2 cardiomyocytes [72]. Conversely, pharmacological iron chelation induced glucose uptake and enhanced insulin sensitivity in HepG2 cells and in the rat liver [28]. These responses were linked to transcriptional induction of the glucose transporter GLUT1 and the insulin receptor InsR following stabilization of HIF1α (hypoxia-inducible factor 1α), which is sensitive to iron-dependent degradation [28]. Iron chelation was also shown to induce GLUT1 expression in rat L6 muscle cells [73]. On the other hand, iron depletion, but also iron overload negatively affected differentiation of human 3T3-L1 adipocytes, underlying the importance of balanced iron metabolism.
Intracellular iron distribution appears to be critical for metabolic functions, at least in some cell types. This became evident with the identification of the outer mitochondrial membrane iron–sulfur cluster protein mitoNEET, a target of the antidiabetic drug pioglitazone [74], as an inhibitor of mitochondrial iron import [75]. Thus, overexpression of mitoNEET preserves insulin sensitivity and reduces oxidative stress in adipocytes, even though it causes expansion of adipose tissue; moreover, it stimulates expression of adiponectin [75]. It appears that mitoNEET, which is stabilized by the antidiabetic drug pioglitazone [74], has pleiotropic functions and is also involved in the control of energy metabolism, iron–sulfur cluster biogenesis and cell proliferation [76]. Interestingly, tetracycline-inducible overexpression of mitoNEET in pancreatic β cells caused glucose intolerance in a mouse model due to activation of a mitophagic pathway [77]. On the other hand, mitoNEET induction in pancreatic α cells elicited protective effects on β cells [77].
Iron is important for proper function of pancreatic β cells (Figure 3); however, excessive iron accumulation has been shown to impair insulin secretion in various settings [35][78][79]. Experiments with rat islets and INS-1E cells identified DMT1 as a key mediator of iron overload and β cell dysfunction following its induction by the inflammatory cytokine IL-1β. Moreover, mice bearing β cell-specific ablation of DMT1 were protected against glucose intolerance in models of type 1 and type 2 diabetes induced by either low-dose streptozotocin treatment or a high-fat diet, respectively [80]. Interestingly, these mice also exhibit defects in glucose-stimulated insulin secretion without inflammatory stimuli, highlighting the importance of physiological iron content in β cell function, but also the critical role of DMT1 in β cell iron supply.
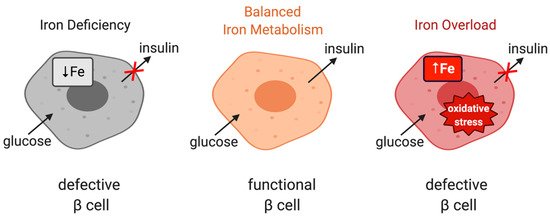
Figure 3. The role of iron in the physiological function of pancreatic β cells. Balanced iron metabolism is critical for proper glucose-stimulated insulin production. Iron deficiency as well as iron overload impair insulin expression.
Surprisingly, mice with genetic hepcidin deficiency (Hamp-/-) [81] or resistance (Slc40a1C326S/C326S) [82] do not exhibit defects in insulin production, in spite of pancreatic iron overload. The reason is that in these mouse models of hemochromatosis, and contrary to human hemochromatosis patients, excess iron accumulates in pancreatic acinar but not β cells. This finding is consistent with the improved insulin sensitivity that has been documented in Hfe-/- mice [61].
Moreover, Hfe-/- mice exhibit improved basal (not insulin-stimulated) glucose uptake in skeletal muscles, in spite of a two-fold increase in iron content of this tissue compared to wild-type controls. This has been attributed to increased phosphorylation of AMPK (AMP-dependent kinase) [83]. However, excessive iron overload appears detrimental for the function of skeletal muscle cells and leads to insulin resistance [84]. Several mechanisms may account for this. Thus, endoplasmic reticulum (ER) stress has been proposed to promote upregulation of TfR1 leading to increased iron uptake and insulin resistance in human skeletal muscle cells, which could be prevented by iron chelation or TfR1 knockdown [85]. Similarly, TfR1-mediated iron overload was shown to be critical for palmitate-induced insulin resistance in the same cell model [86]. Autophagy plays a crucial role in skeletal muscles’ response to insulin via the Akt pathway [87]. In this context, iron overload was observed to perturb insulin signaling by impairing autophagy processes [88].
Interestingly, there appears to be distinct cellular repercussions between acute and prolonged exposures to iron overload. Hence, short-term iron overload stimulated autophagy induction through inhibition of the mammalian target of rapamycin complex 1 protein (mTORC1), conceivably as a compensatory measure against iron overload-related stress. Optimal autophagic flux, however, requires proper fusion of autophagosomes with lysosomes to form autolysosomes. Following degradation of autophagic materials, autolysosomal membranes must be resorbed into the endomembrane system in order to regenerate functional lysosomes, a newly discovered process known as autophagic-lysosome regeneration (ALR) [89]. Chronic iron overload eventually impaired ALR, causing accumulation of autolysosome while depleting free lysosomes and effectively halting autophagic flux [88]. This effect was mediated by the loss of mTORC1 reactivation, followed by insulin resistance and decreased Akt phosphorylation. Both ALR and insulin sensitivity were recovered upon iron withdrawal. Furthermore, forced mTORC1 activation not only prevented autolysosome accumulation but also recovered autophagic flux and insulin sensitivity under iron overload. This evidence suggests that skeletal muscle iron overload specifically induces insulin resistance by inhibiting ALR and autophagic flux.
6. Conclusions
Both iron and glucose are essential for cellular energy production: the former as a component of key metabolic enzymes and the latter as the principal energy source. Clinical studies suggested that deregulation of iron metabolism in iron overload disorders is associated with metabolic dysfunction. Moreover, deregulation of glucose homeostasis in the metabolic syndrome often correlates with iron overload. Nevertheless, attempts to target iron pathways in order to improve metabolic functions have had limited success thus far. This may be related to a knowledge gap on mechanisms linking iron and glucose homeostasis. Herein, we presented data, mainly obtained from mouse models, suggesting an interplay between systemic iron and glucose homeostasis involving their hormonal regulators hepcidin and insulin, respectively. A deeper understanding of the molecular mechanisms linking iron and glucose metabolism may pave the way for the identification of new pharmacological targets and the development of relevant therapeutic interventions for the treatment of common metabolic disorders.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21207773
References
- Frausto da Silva, J.J.R.; Williams, R.J.P. The Biological Chemistry of the Elements. The Inorganic Chemistry of Life; Clarendon Press: Oxford, UK, 1991; pp. 319–369.
- Oexle, H.; Gnaiger, E.; Weiss, G. Iron-dependent changes in cellular energy metabolism: Influence on citric acid cycle and oxidative phosphorylation. Biochim. Biophys. Acta 1999, 1413, 99–107.
- Volani, C.; Doerrier, C.; Demetz, E.; Haschka, D.; Paglia, G.; Lavdas, A.A.; Gnaiger, E.; Weiss, G. Dietary iron loading negatively affects liver mitochondrial function. Metallomics 2017, 9, 1634–1644.
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell. Res. 2019, 1866, 118535.
- Katsarou, A.; Pantopoulos, K. Basics and principles of cellular and systemic iron homeostasis. Mol. Asp. Med. 2020.
- Lehmann, A.; Hornby, P.J. Intestinal SGLT1 in metabolic health and disease. Am. J. Physiol. Liver Physiol. 2016, 310, G887–G898.
- Kellett, G.L.; Brot-Laroche, E. Apical GLUT2: A Major Pathway of Intestinal Sugar Absorption. Diabetes 2005, 54, 3056–3062.
- Navale, A.M.; Paranjape, A.N. Glucose transporters: Physiological and pathological roles. Biophys. Rev. 2016, 8, 5–9.
- McCulloch, L.J.; Van De Bunt, M.; Braun, M.; Frayn, K.N.; Clark, A.; Gloyn, A.L. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: Implications for understanding genetic association signals at this locus. Mol. Genet. Metab. 2011, 104, 648–653.
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223.
- Simcox, J.A.; McClain, D.A. Iron and Diabetes Risk. Cell Metab. 2013, 17, 329–341.
- Fernández-Real, J.M.; Manco, M. Effects of iron overload on chronic metabolic diseases. Lancet Diabetes Endocrinol. 2014, 2, 513–526.
- Utzschneider, K.M.; Kowdley, K.V. Hereditary hemochromatosis and diabetes mellitus: Implications for clinical practice. Nat. Rev. Endocrinol. 2010, 6, 26–33.
- De Sanctis, V.; Soliman, A.T.; Elsedfy, H.; Pepe, A.; Kattamis, C.; El Kholy, M.; Yassin, M. Diabetes and Glucose Metabolism in Thalassemia Major: An Update. Expert Rev. Hematol. 2016, 9, 401–408.
- Bozzini, C.; Girelli, D.; Olivieri, O.; Martinelli, N.; Bassi, A.; De Matteis, G.; Tenuti, I.; Lotto, V.; Friso, S.; Pizzolo, F.; et al. Prevalence of Body Iron Excess in the Metabolic Syndrome. Diabetes Care 2005, 28, 2061–2063.
- Fernández-Real, J.M.; Ricart, W.; Arroyo, E.; Balançá, R.; Casamitjana-Abella, R.; Cabrero, D.; Fernández-Castañer, M.; Soler, J. Serum ferritin as a component of the insulin resistance syndrome. Diabetes Care 1998, 21, 62–68.
- Lee, D.H.; Liu, D.Y.; Jacobs, D.R., Jr.; Shin, H.R.; Song, K.; Lee, I.K.; Kim, B.; Hider, R.C. Common presence of non-transferrin-bound iron among patients with type 2 diabetes. Diabetes Care 2006, 29, 1090–1095.
- Mendler, M.-H.; Turlin, B.; Moirand, R.; Jouanolle, A.-M.; Sapey, T.; Guyader, D.; Le Gall, J.-Y.; Brissot, P.; David, V.; Deugnier, Y. Insulin resistance–associated hepatic iron overload. Gastroenterology 1999, 117, 1155–1163.
- Deugnier, Y.; Bardou-Jacquet, E.; Lainé, F. Dysmetabolic iron overload syndrome (DIOS). Presse Méd. 2017, 46, e306–e311.
- Aigner, E.; Feldman, A.; Datz, C. Obesity as an Emerging Risk Factor for Iron Deficiency. Nutrients 2014, 6, 3587–3600.
- Bellentani, S. The epidemiology of non-alcoholic fatty liver disease. Liver Int. 2017, 37, 81–84.
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20.
- Corradini, E.; Pietrangelo, A. Iron and steatohepatitis. J. Gastroenterol. Hepatol. 2012, 27, 42–46.
- Sorrentino, P.; D’Angelo, S.; Ferbo, U.; Micheli, P.; Bracigliano, A.; Vecchione, R. Liver iron excess in patients with hepatocellular carcinoma developed on non-alcoholic steato-hepatitis. J. Hepatol. 2009, 50, 351–357.
- Dongiovanni, P.; Fracanzani, A.L.; Fargion, S.; Valenti, L. Iron in fatty liver and in the metabolic syndrome: A promising therapeutic target. J. Hepatol. 2011, 55, 920–932.
- Valenti, L.; Fracanzani, A.L.; Dongiovanni, P.; Bugianesi, E.; Marchesini, G.; Manzini, P.; Vanni, E.; Fargion, S. Iron Depletion by Phlebotomy Improves Insulin Resistance in Patients With Nonalcoholic Fatty Liver Disease and Hyperferritinemia: Evidence from a Case-Control Study. Am. J. Gastroenterol. 2007, 102, 1251–1258.
- Valenti, L.; Fracanzani, A.L.; Dongiovanni, P.; Rovida, S.; Rametta, R.; Fatta, E.; Pulixi, E.A.; Maggioni, M.; Fargion, S. A randomized trial of iron depletion in patients with nonalcoholic fatty liver disease and hyperferritinemia. World J. Gastroenterol. 2014, 20, 3002–3010.
- Dongiovanni, P.; Valenti, L.; Fracanzani, A.L.; Gatti, S.; Cairo, G.; Fargion, S. Iron Depletion by Deferoxamine Up-Regulates Glucose Uptake and Insulin Signaling in Hepatoma Cells and in Rat Liver. Am. J. Pathol. 2008, 172, 738–747.
- Cooksey, R.C.; Jones, D.; Gabrielsen, S.; Huang, J.; Simcox, J.A.; Luo, B.; Soesanto, Y.; Rienhoff, H.; Abel, E.D.; McClain, D.A. Dietary iron restriction or iron chelation protects from diabetes and loss of beta-cell function in the obese (ob/ob lep-/-) mouse. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1236–E1243.
- Murali, A.R.; Gupta, A.; Brown, K.E. Systematic review and meta-analysis to determine the impact of iron depletion in dysmetabolic iron overload syndrome and non-alcoholic fatty liver disease. Hepatol. Res. 2017, 48, E30–E41.
- Pietrangelo, A. Hemochromatosis: An endocrine liver disease. Hepatology 2007, 46, 1291–1301.
- Wang, H.; Li, H.; Jiang, X.; Shi, W.; Shen, Z.; Li, M. Hepcidin Is Directly Regulated by Insulin and Plays an Important Role in Iron Overload in Streptozotocin-Induced Diabetic Rats. Diabetes 2014, 63, 1506–1518.
- Aigner, E.; Felder, T.K.; Oberkofler, H.; Hahne, P.; Auer, S.; Soyal, S.; Stadlmayr, A.; Schwenoha, K.; Pirich, C.; Hengster, P.; et al. Glucose acts as a regulator of serum iron by increasing serum hepcidin concentrations. J. Nutr. Biochem. 2013, 24, 112–117.
- Mao, X.; Chen, H.; Tang, J.; Wang, L.; Shu, T. Hepcidin links gluco-toxicity to pancreatic beta cell dysfunction by inhibiting Pdx-1 expression. Endocr. Connect. 2017, 6, 121–128.
- Shu, T.; Lv, Z.; Xie, Y.; Tang, J.; Mao, X. Hepcidin as a key iron regulator mediates glucotoxicity-induced pancreatic beta-cell dysfunction. Endocr. Connect. 2019, 8, 150–161.
- Tajima, S.; Ikeda, Y.; Sawada, K.; Yamano, N.; Horinouchi, Y.; Kihira, Y.; Ishizawa, K.; Izawa-Ishizawa, Y.; Kawazoe, K.; Tomita, S.; et al. Iron reduction by deferoxamine leads to amelioration of adiposity via the regulation of oxidative stress and inflammation in obese and type 2 diabetes KKAy mice. Am. J. Physiol. Metab. 2012, 302, E77–E86.
- Vecchi, C.; Montosi, G.; Garuti, C.; Corradini, E.; Sabelli, M.; Canali, S.; Pietrangelo, A. Gluconeogenic Signals Regulate Iron Homeostasis via Hepcidin in Mice. Gastroenterology 2014, 146, 1060–1069.e3.
- Chung, B.; Matak, P.; McKie, A.T.; Sharp, P.A. Leptin Increases the Expression of the Iron Regulatory Hormone Hepcidin in HuH7 Human Hepatoma Cells. J. Nutr. 2007, 137, 2366–2370.
- Yamamoto, K.; Kuragano, T.; Kimura, T.; Nanami, M.; Hasuike, Y.; Nakanishi, T. Interplay of adipocyte and hepatocyte: Leptin upregulates hepcidin. Biochem. Biophys. Res. Commun. 2018, 495, 1548–1554.
- Altamura, S.; Kopf, S.; Schmidt, J.; Müdder, K.; Da Silva, A.R.; Nawroth, P.; Muckenthaler, M.U. Uncoupled iron homeostasis in type 2 diabetes mellitus. J. Mol. Med. 2017, 95, 1387–1398.
- Padda, R.S.; Gkouvatsos, K.; Guido, M.; Mui, J.; Vali, H.; Pantopoulos, K. A high-fat diet modulates iron metabolism but does not promote liver fibrosis in hemochromatotic Hjv-/- mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G251–G261.
- Chung, J.; Kim, M.S.; Han, S.N. Diet-induced obesity leads to decreased hepatic iron storage in mice. Nutr. Res. 2011, 31, 915–921.
- Sonnweber, T.; Ress, C.; Nairz, M.; Theurl, I.; Schroll, A.; Murphy, A.T.; Wroblewski, V.; Witcher, D.R.; Moser, P.; Ebenbichler, C.; et al. High-fat diet causes iron deficiency via hepcidin-independent reduction of duodenal iron absorption. J. Nutr. Biochem. 2012, 23, 1600–1608.
- Meli, R.; Raso, G.M.; Irace, C.; Simeoli, R.; Di Pascale, A.; Paciello, O.; Pagano, T.B.; Calignano, A.; Colonna, A.; Santamaria, R. High Fat Diet Induces Liver Steatosis and Early Dysregulation of Iron Metabolism in Rats. PLoS ONE 2013, 8, e66570.
- Dongiovanni, P.; Lanti, C.; Gatti, S.; Rametta, R.; Recalcati, S.; Maggioni, M.; Fracanzani, A.L.; Riso, P.; Cairo, G.; Fargion, S.; et al. High Fat Diet Subverts Hepatocellular Iron Uptake Determining Dysmetabolic Iron Overload. PLoS ONE 2015, 10, e0116855.
- Engin, A. The Pathogenesis of Obesity-Associated Adipose Tissue Inflammation. Adv. Exp. Med. Biol. 2017, 960, 221–245.
- Coimbra, S.; Catarino, C.; Santos-Silva, A. The role of adipocytes in the modulation of iron metabolism in obesity. Obes. Rev. 2013, 14, 771–779.
- Folgueras, A.R.; Martin de Lara, F.; Pendas, A.M.; Garabaya, C.; Rodriguez, F.; Astudillo, A.; Bernal, T.; Cabanillas, R.; Lopez-Otin, C.; Velasco, G. The membrane-bound serine protease matriptase-2 (Tmprss6) is an essential regulator of iron homeostasis. Blood 2008, 112, 2539–2545.
- Folgueras, A.R.; Freitas-Rodríguez, S.; Ramsay, A.J.; Garabaya, C.; Rodríguez, F.; Velasco, G.; López-Otín, C. Matriptase-2 deficiency protects from obesity by modulating iron homeostasis. Nat. Commun. 2018, 9, 1350.
- Saad, M.J.A.; Santos, A.; Prada, P.O. Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiology 2016, 31, 283–293.
- Zmora, N.; Bashiardes, S.; Levy, M.; Elinav, E. The Role of the Immune System in Metabolic Health and Disease. Cell Metab. 2017, 25, 506–521.
- Tilg, H.; Cani, P.D.; Mayer, E.A. Gut microbiome and liver diseases. Gut 2016, 65, 2035–2044.
- Yilmaz, B.; Li, H. Gut Microbiota and Iron: The Crucial Actors in Health and Disease. Pharmaceuticals 2018, 11, 98.
- Lee, T.; Clavel, T.; Smirnov, K.; Schmidt, A.; Lagkouvardos, I.; Walker, A.; Lucio, M.; Michalke, B.; Schmitt-Kopplin, P.; Fedorak, R.; et al. Oral versus intravenous iron replacement therapy distinctly alters the gut microbiota and metabolome in patients with IBD. Gut 2016, 66, 863–871.
- La Carpia, F.; Wojczyk, B.; Rebbaa, A.; Tang, A.; Hod, E.A. Chronic Transfusion and Iron Overload Modify the Mouse Gut Microbiome. Blood 2016, 128, 200.
- Buhnik-Rosenblau, K.; Moshe-Belizowski, S.; Danin-Poleg, Y.; Meyron-Holtz, E.G. Genetic modification of iron metabolism in mice affects the gut microbiota. BioMetals 2012, 25, 883–892.
- Das, N.K.; Schwartz, A.J.; Barthel, G.; Inohara, N.; Liu, Q.; Sankar, A.; Hill, D.R.; Ma, X.; Lamberg, O.; Schnizlein, M.K.; et al. Microbial Metabolite Signaling Is Required for Systemic Iron Homeostasis. Cell Metab. 2020, 31, 115–130.e6.
- Dongiovanni, P.; Ruscica, M.; Rametta, R.; Recalcati, S.; Steffani, L.; Gatti, S.; Girelli, D.; Cairo, G.; Magni, P.; Fargion, S.; et al. Dietary Iron Overload Induces Visceral Adipose Tissue Insulin Resistance. Am. J. Pathol. 2013, 182, 2254–2263.
- Fernández-Real, J.M.; Moreno-Navarrete, J.M.; Ricart, W. Circulating Retinol-Binding Protein-4 Concentration Might Reflect Insulin Resistance–Associated Iron Overload. Diabetes 2008, 57, 1918–1925.
- Gao, Y.; Li, Z.; Gabrielsen, J.S.; Simcox, J.A.; Lee, S.-H.; Jones, D.; Cooksey, B.; Stoddard, G.; Cefalu, W.T.; McClain, D.A. Adipocyte iron regulates leptin and food intake. J. Clin. Investig. 2015, 125, 3681–3691.
- Gabrielsen, J.S.; Gao, Y.; Simcox, J.A.; Huang, J.; Thorup, D.; Jones, D.; Cooksey, R.C.; Gabrielsen, D.; Adams, T.D.; Hunt, S.C.; et al. Adipocyte iron regulates adiponectin and insulin sensitivity. J. Clin. Investig. 2012, 122, 3529–3540.
- Britton, L.; Jaskowski, L.-A.; Bridle, K.; Secondes, E.; Wallace, D.F.; Santrampurwala, N.; Reiling, J.; Miller, G.; Mangiafico, S.; Andrikopoulos, S.; et al. Ferroportin Expression in Adipocytes Does Not Contribute to Iron Homeostasis or Metabolic Responses to a High Calorie Diet. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 319–331.
- Miranda, M.A.; Pierre, C.L.S.; Macias-Velasco, J.F.; Nguyen, H.A.; Schmidt, H.; Agnello, L.T.; Wayhart, J.P.; Lawson, H.A. Dietary iron interacts with genetic background to influence glucose homeostasis. Nutr. Metab. 2019, 16, 13.
- Bekri, S.; Gual, P.; Anty, R.; Luciani, N.; Dahman, M.; Ramesh, B.; Iannelli, A.; Staccini–Myx, A.; Casanova, D.; Ben Amor, I.; et al. Increased Adipose Tissue Expression of Hepcidin in Severe Obesity Is Independent From Diabetes and NASH. Gastroenterology 2006, 131, 788–796.
- Gotardo, E.M.F.; Dos Santos, A.N.; Miyashiro, R.A.; Gambero, S.; Rocha, T.; Ribeiro, M.L.; Gambero, A. Mice That Are Fed a High-Fat Diet Display Increased Hepcidin Expression in Adipose Tissue. J. Nutr. Sci. Vitaminol. 2013, 59, 454–461.
- Orr, J.S.; Kennedy, A.; Anderson-Baucum, E.K.; Webb, C.D.; Fordahl, S.C.; Erikson, K.M.; Zhang, Y.; Etzerodt, A.; Moestrup, S.K.; Hasty, A.H. Obesity Alters Adipose Tissue Macrophage Iron Content and Tissue Iron Distribution. Diabetes 2014, 63, 421–432.
- Hubler, M.J.; Peterson, K.R.; Hasty, A.H. Iron homeostasis: A new job for macrophages in adipose tissue? Trends Endocrinol. Metab. 2015, 26, 101–109.
- Davis, R.J.; Corvera, S.; Czech, M.P. Insulin stimulates cellular iron uptake and causes the redistribution of intracellular transferrin receptors to the plasma membrane. J. Biol. Chem. 1986, 261, 8708–8711.
- Biswas, S.; Tapryal, N.; Mukherjee, R.; Kumar, R.; Mukhopadhyay, C.K. Insulin promotes iron uptake in human hepatic cell by regulating transferrin receptor-1 transcription mediated by hypoxia inducible factor-1. Biochim. Biophys. Acta 2013, 1832, 293–301.
- Green, A.; Basile, R.; Rumberger, J.M. Transferrin and iron induce insulin resistance of glucose transport in adipocytes. Metabolism 2006, 55, 1042–1045.
- Varghese, J.; James, J.; Vaulont, S.; McKie, A.; Jacob, M. Increased intracellular iron in mouse primary hepatocytes in vitro causes activation of the Akt pathway but decreases its response to insulin. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1870–1882.
- Sung, H.K.; Song, E.; Jahng, J.W.S.; Pantopoulos, K.; Sweeney, G. Iron induces insulin resistance in cardiomyocytes via regulation of oxidative stress. Sci. Rep. 2019, 9, 4668.
- Potashnik, R.; Kozlovsky, N.; Ben-Ezra, S.; Rudich, A.; Bashan, N. Regulation of glucose transport and GLUT-1 expression by iron chelators in muscle cells in culture. Am. J. Physiol. Metab. 1995, 269, E1052–E1058.
- Paddock, M.L.; E Wiley, S.; Axelrod, H.L.; Cohen, A.E.; Roy, M.; Abresch, E.C.; Capraro, M.; Murphy, A.N.; Nechushtai, R.; Dixon, J.E.; et al. MitoNEET is a uniquely folded 2Fe 2S outer mitochondrial membrane protein stabilized by pioglitazone. Proc. Natl. Acad. Sci. USA 2007, 104, 14342–14347.
- Kusminski, C.M.; Holland, W.L.; Sun, K.; Park, J.; Spurgin, S.B.; Lin, Y.; Askew, G.R.; Simcox, J.A.; McClain, D.A.; Li, C.; et al. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat. Med. 2012, 18, 1539–1549.
- Mittler, R.; Darash-Yahana, M.; Sohn, Y.S.; Bai, F.; Song, L.; Cabantchik, I.Z.; Jennings, P.A.; Onuchic, J.N.; Nechushtai, R. NEET Proteins: A New Link Between Iron Metabolism, Reactive Oxygen Species, and Cancer. Antioxidants Redox Signal. 2019, 30, 1083–1095.
- Kusminski, C.M.; Chen, S.; Ye, R.; Sun, K.; Wang, Q.A.; Spurgin, S.B.; Sanders, P.E.; Brozinick, J.T.; Geldenhuys, W.J.; Li, W.H.; et al. MitoNEET-Parkin Effects in Pancreatic alpha- and beta-Cells, Cellular Survival, and Intrainsular Cross Talk. Diabetes 2016, 65, 1534–1555.
- Cooksey, R.C.; Jouihan, H.A.; Ajioka, R.S.; Hazel, M.W.; Weiss, S.M.; Kushner, J.P.; McClain, D.A. Oxidative Stress, β-Cell Apoptosis, and Decreased Insulin Secretory Capacity in Mouse Models of Hemochromatosis. Endocrinology 2004, 145, 5305–5312.
- Masuda, Y.; Ichii, H.; Vaziri, N.D. At pharmacologically relevant concentrations intravenous iron preparations cause pancreatic beta cell death. Am. J. Transl. Res. 2013, 6, 64–70.
- Hansen, J.B.; Tonnesen, M.F.; Madsen, A.N.; Hagedorn, P.H.; Friberg, J.; Grunnet, L.G.; Heller, R.S.; Nielsen, A.O.; Storling, J.; Baeyens, L.; et al. Divalent metal transporter 1 regulates iron-mediated ROS and pancreatic beta cell fate in response to cytokines. Cell Metab. 2012, 16, 449–461.
- Ramey, G.; Faye, A.; Durel, B.; Viollet, B.; Vaulont, S. Iron overload in Hepc1(-/-) mice is not impairing glucose homeostasis. FEBS Lett. 2007, 581, 1053–1057.
- Altamura, S.; Kessler, R.; Gröne, H.-J.; Gretz, N.; Hentze, M.W.; Galy, B.; Muckenthaler, M.U. Resistance of Ferroportin to Hepcidin Binding causes Exocrine Pancreatic Failure and Fatal Iron Overload. Cell Metab. 2014, 20, 359–367.
- Huang, J.; Gabrielsen, J.S.; Cooksey, R.C.; Luo, B.; Boros, L.G.; Jones, D.; Jouihan, H.A.; Soesanto, Y.; Knecht, L.; Hazel, M.W.; et al. Increased Glucose Disposal and AMP-dependent Kinase Signaling in a Mouse Model of Hemochromatosis. J. Biol. Chem. 2007, 282, 37501–37507.
- DeFronzo, R.A.; Tripathy, D. Skeletal Muscle Insulin Resistance Is the Primary Defect in Type 2 Diabetes. Diabetes Care 2009, 32, S157–S163.
- Lee, K.; Cui, R.; Kim, D.J.; Choi, S.; Lee, W.J.; Kang, Y.; Kim, T.H.; Moon, H.U.; Jeon, J.Y.; Han, S.J.; et al. Iron Overload Plays an Important Role in ER Stress-Induced Insulin Resistance in Human Skeletal Muscle Cells. Diabetes 2018, 67, 1912.
- Cui, R.; Choi, S.-E.; Kim, T.H.; Lee, H.J.; Lee, S.J.; Kang, Y.; Jeon, J.Y.; Kim, H.J.; Lee, K.-W. Iron overload by transferrin receptor protein 1 regulation plays an important role in palmitate-induced insulin resistance in human skeletal muscle cells. FASEB J. 2018, 33, 1771–1786.
- Ahlstrom, P.; Rai, E.; Chakma, S.; Cho, H.H.; Rengasamy, P.; Sweeney, G. Adiponectin improves insulin sensitivity via activation of autophagic flux. J. Mol. Endocrinol. 2017, 59, 339–350.
- Jahng, J.W.S.; Alsaadi, R.M.; Palanivel, R.; Song, E.; Hipolito, V.E.B.; Sung, H.K.; Botelho, R.J.; Russell, R.C.; Sweeney, G. Iron overload inhibits late stage autophagic flux leading to insulin resistance. EMBO Rep. 2019, 20, e47911.
- Chen, Y.; Yu, L. Development of Research into Autophagic Lysosome Reformation. Mol. Cells 2018, 41, 45–49.