9.1. Tregs
Tregs were first reported by Sakaguchi et al. in thymectomized mice [
57]. They consist of 5–10% of CD4+ T cells and are broadly classified as thymic-derived and peripherally induced Tregs [
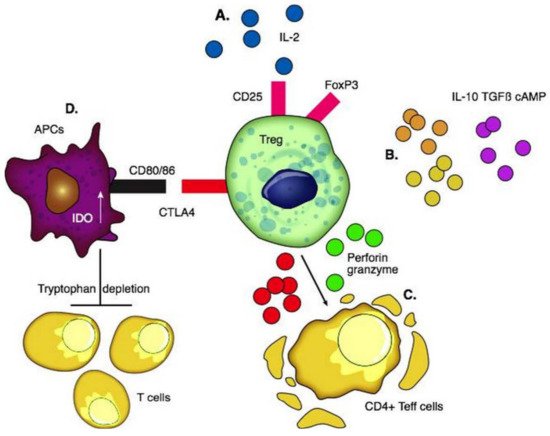
58]. CD4, FoxP3, high levels of CD25, and low levels of CD127 are expressed on the surface. The CD25 marker serves as an “IL-2 sink” and consumes IL-2 which is the key cytokine required for the differentiation and maintenance of Tregs and stabilization of FoxP3 expression. The preferential consumption of IL-2 by Tregs suppresses the expansion of effector T cells [
59]. Once activated, Tregs travel to the site of inflammation and prevent collateral tissue destruction by CD4+ and CD8+ T cells. Tregs secrete IL-10 [
60], IL-35 [
60], TGF β [
61], and cAMP [
62] to create an immunotolerant environment. Through the secretion of granzymes and perforins, they cause apoptosis of effector T cells [
63,
64]. CTLA4 is an essential negative regulator of T cell response on the surface of Tregs; the interaction between CTLA4 and costimulatory CD80/86 ligands on the surface of APCs activates indoleamine dioxygenase (IDO) in APCs. IDO depletes tryptophan in the local environment and consequently inhibits T effector cells [
65] ().
Figure 1. Functions of regulatory T cells. (A) Interaction of pro-inflammatory IL-2 with CD25 for the differentiation and maintenance of Tregs and stabilization of FoxP3. (B) Production of anti-inflammatory cytokines including IL-10, TGFβ, and cAMP to create an immunotolerant environment. (C) Secretion of granzymes and perforins causes apoptosis of CD4+T effector cells. (D) CTLA4 interacts with co-stimulatory CD80 and CD86 ligands on the surface of APCs and activates IDO in them which depletes tryptophan in the local environment and consequently inhibits the proliferation of T cells.
Tregs control transplant rejection by first migrating to the graft to induce graft tolerance and then drain into lymph nodes and help maintain tolerance [
66]. Two types of Tregs can be expanded for clinical use: antigen-specific Tregs and polyclonal Tregs. On one hand, antigen-specific Tregs have specificity to one antigen and suppress effector T cells of distinct antigenic specificity making them more effective [
67]. On the other hand, polyclonal Tregs are easier to manufacture [
67]. Interestingly, Tregs demonstrate “infectious tolerance” [
68], where the suppressive capacity of Tregs is transferred and can lead to the generation and expansion of a new population of antigen-specific Tregs distinct from the original Treg population. This occurs through the production of cytokines which induces the conversion of naïve T cells into Tregs [
17]. Thus, tolerance persists much longer than the longevity of Tregs used.
The first clinical trial of T-cell-based therapy to induce OT was conducted by Todo et al., using an ex vivo-generated regulatory T-cell-enriched cell product in 10 adult patients post-LT [
69]. The cultured Treg-enriched product significantly inhibited the proliferation of recipient lymphocytes in an ex-vivo Mixed Lymphocyte Reaction assay. In the human trial, the Treg product was administered on post-op day 13 along with a standard IS regimen including steroids, mycophenolate mofetil, and tacrolimus. IS drugs were gradually discontinued over 18 months. All 10 recipients maintained stable graft function. Seven patients with non-immunological liver diseases successfully achieved weaning and did not require ISs between 16 and 33 months. The other three recipients with autoimmune liver diseases developed mild rejection during weaning and were resumed on IS.
King’s group [
23] proved the safety, applicability, and biological activity of autologous Treg adoptive transfer in humans in cadaveric liver transplantation. Circulating polyclonal Tregs were isolated from the recipient’s blood and expanded ex-vivo using IL-2 and rapamycin [
70]. In three patients, Tregs were harvested pre-transplant, standard IS was administered for three months post-transplant, a biopsy was performed to exclude subclinical allograft rejection, and then Tregs were infused. In six patients, Tregs were harvested 8–14 months after transplant, with simultaneous liver biopsy, and the patients received expanded Treg infusion two months later. In recipients who received a single dose of 4.5 million Tregs/kg, a gradual decrease in T cell responses directed against donor cells was observed. Although they were not successful in weaning patients off IS, the development of donor-specific hyporesponsiveness is considered one of the hallmarks of transplantation tolerance [
23].
Tregs demonstrate promise for targeted allograft tolerance and an alternative to classical IS. The first successful pilot study by Todo led to multiple OT induction trials currently using Treg cell therapy in LT [
17]. Tregs do not persist in large numbers in the circulation after infusion due to low IL-2 availability. Even if they persist, a study revealed that Tregs may acquire a pro-inflammatory T effector phenotype as a consequence of FoxP3 instability [
71]. Hence, their long-term viability needs to be addressed. In addition, due to the high frequency of alloreactive T effector cells post-transplant, Treg administration alone is insufficient and a combined regimen of lymphodepletion and donor-alloantigen-reactive Tregs is most effective in long-term graft survival [
72]. Chimeric antigen receptor (CAR) T cells could be a promising method for the production of antigen-specific Tregs over polyclonal Tregs [
73]. These engineered Tregs have a superior allospecific immune response and proliferation capacity [
73]. Noyon et al. successfully transduced natural Tregs to donor-specific CAR Tregs without altering their regulatory phenotype. These CAR Tregs were far more potent in preventing rejection after a skin transplant in a mouse and could be applied to produce potent donor-antigen-specific Tregs after LT [
73].
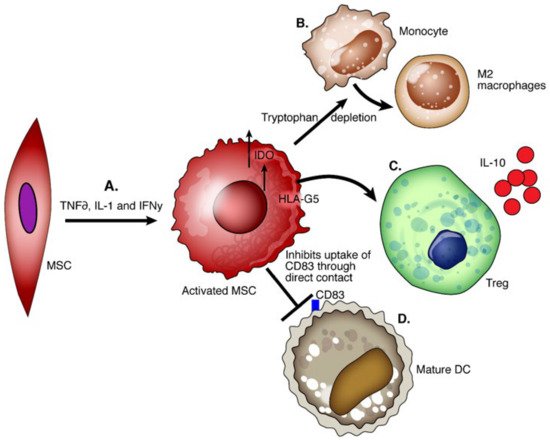
9.2. DCregs
DCs are stellate-shaped APCs expressing CD11c and CD1a. They arise from the bone marrow and seed into peripheral tissue. Mature DCs produce pro-inflammatory cytokines, while liver-derived DCs have a lower maturity status and produce high levels of regulatory factors including IL-10, IL-27, retinoic acid, and prostaglandin E2 [
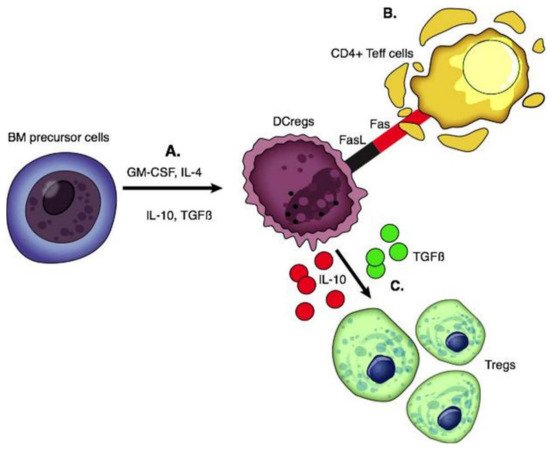
74]. DCs with immune tolerance properties are called DCregs; these cells express high levels of MHC, T cell co-inhibitory ligands (PDL-1), and death-inducing ligand (FasL) with low expression of co-stimulatory molecules [
75]. DCregs are immature cells that induce apoptosis of alloantigen-specific T cells through the Fas/FasL pathway and IDO and generate Tregs and Bregs through the production of IL-10 and TGF β [
76,
77] ().
Figure 2. Functions of regulatory dendritic cells. (A) DCregs are generated from BM precursor cells by adding GM-CSF with IL-4, IL-10, and TGFβ. (B) DCregs express FasL and interact with the Fas on activated CD4 T effector cells which is associated with T cell apoptosis. (C) Production of IL-10 and TGFβ leads to the generation of Tregs.
In vitro generation of DCregs has been achieved by utilizing cytokines (IL-10, IL-4, TGF-β, and VEGF), pharmaceutical agents (aspirin, PGE2, histamine, β2 agonists, corticosteroids), and IS (cyclosporin A, rapamycin, and mycophenolate mofetil) [
78]. The first and only clinical trial testing the efficacy of a single infusion of donor-derived DCregs in LT recipients is currently in phase I/II [
79,
80]. Monocytes from prospective liver donors were cryopreserved, then thawed and treated with L-glutamine, GM-CSF, IL-4, Vitamin D3, and IL-10. DCregs in this study exhibited a tolerogenic profile of high PDL1 and IL-10, resistance to maturation, and potential to modulate alloreactive T cell responses. Fifteen live donor LT donor-recipient pairs were recruited between 2017 and 2020 at the University of Pittsburgh and received a single infusion of these donor-derived DCregs one week prior to transplantation [
79,
80]. The timing of infusion of donor-derived DCs seven days prior to transplant has been supported by many prior studies; this timing is based on the hypothesis that DCs are most involved in the very early stages of the immune response and hyperacute rejection [
38,
81,
82,
83]. DCreg infusion in transplantation has resulted in decreased memory CD8+ T cells and increased Tregs in circulation [
80]. This effective blunting of memory cells is a major victory in the promotion of long-term allograft survival [
84]. Intact donor-derived DCregs with preserved regulatory phenotype were detected in the circulation shortly after their infusion, but there was no long-term persistence of donor DCregs. The Pittsburgh Study (NCT03164265) is yet to report on the post-transplant clinical and immunological outcomes, and whether this approach to donor DCreg therapy could result in the enhanced success of early IS withdrawal [
80]. The estimated completion date of this study is June 2023.
Of note, ex-vivo expansion of DCregs yielded high numbers of viable cells, constituting a possible advantage over other types and sources of regulatory cells (e.g., autologous Tregs) which require several weeks to expand. Currently, donor-derived DCregs are restricted to live-donor organ transplantation and alternative therapies such as autologous DCregs loaded with donor antigen are promising for deceased donors [
85]. Genetically engineered DCs may be a feasible approach to improve the therapy of allograft rejection as DCs can be virally transduced to express various immunosuppressive molecules, such as IL-10 and TGFβ, to further enhance their tolerogenic potential [
86].