1.Skin Pigmentation
Skin color is one of the topmost concerns for human beings as it provides uniformity, identity and represents the overall appearance of a person [
1]. Production, quantity, quality, and distribution of melanin, a group of natural pigments, determines the color of human skin, eyes, and hair. It is crucially involved in the defense mechanism to protect skin from adverse effects of ultraviolet radiation (UVR) and oxidative stress of environmental pollutants [
2]. Synthesis of melanin starts with formation of melanosomes, melanin-containing organelles, in epidermal melanocytes by a process called melanogenesis [
3,
4]. Melanocytes are specialized cells derived from unpigmented precursor cells called melanoblasts, originating from embryonic neural crest cells which can migrate towards the skin and other tissues during embryogenesis [
5,
6,
7,
8]. Skin pigmentation is a specific and complex mechanism that occurs due to accumulation of melanosomes in keratinocytes to protect skin from solar irradiation. Melanin pigment can be divided into two types—pheomelanin and eumelanin. Pheomelanin has a reddish to brownish color, causing reactive oxygen species production under UVR stimulation. Eumelanin has black color, which works mainly to protect the nucleus from UVR. Skin color differences can emerge from the quantity and quality (pheo/eumelanin ratio) of melanin produced as well as size, number, composition, mode of transfer, distribution, and degradation of melanosomes inside keratinocytes, whereas melanocytes numbers typically remain relatively constant [
8]. Keratinocytes have a significant role in regulating adhesion, proliferation, survival, and morphology of melanocytes [
8].
There are various nonspecific intrinsic factors (hormonal environment, inflammation) and extrinsic factors (solar irradiation such as ultraviolet irradiation, environmental pollution, drugs) modulating genetically determined melanin levels. Various stimuli including skin eruptions such as eczema, allergens, microbial infection, pathogens, chemical stimuli and physical damage, ultraviolet light exposure, and aging can all lead to skin inflammation [
9,
10,
11,
12], defined as a defensive reaction of living tissues with a vascular system in response to exogenous and endogenous stimuli [
13]. In dermatology and cosmetology, hyperpigmentation or hypopigmentation after inflammation is a major problem and commonly seen due to acute or chronic inflammatory skin reactions [
14]. Inflammatory mediators are chemical factors involved in mediating inflammatory reactions and mainly secreted by T helper cells (Th), monocytes and macrophages, dendritic cells, as well as epidermal keratinocytes. Recent studies have revealed a close relationship between inflammatory cytokines and skin pigmentation. Diversified interleukins (ILs) and varieties of inflammatory mediators can also participate in melanogenesis regulation by modulating proliferation and differentiation of human epidermal melanocytes, and also promote or inhibit melanogenesis-related gene expression directly or indirectly [
14,
15,
16]. Many genes are involved in regulating pigmentation at various levels, as well as mutations in several cause pigmentary disorders.
4. Inflammatory Cytokines Induce Melanogenesis
Several studies have observed the involvement of local inflammatory factors in skin pigmentation regulation, which are presented in (). It has been revealed that IL-4 directly takes part in inhibiting melanogenesis in normal human epidermal melanocytes (NHEMs) by downregulating MITF, TYRP1, and TYRP2 expression through the JAK2-STAT6 signaling pathway [
16]. IL-4 is mainly produced by Th2 cells. In chronic inflammation, basophils, eosinophils, and mast cells can also secrete IL-4 [
97,
98]. IL-4 plays a crucial role in IgE generation in hypersensitivity, inflammation induction, autoimmunity [
99] and is also involved in the maintenance of Th2 lymphocytes and acts as an autocrine growth factor of differentiated Th2 cells [
100]. It is hypothesized that skewed balance to Th1 is directly responsible for vitiligo development by changing IFN-γ/IL-4, Tbet/Gata3 profiles in vitiligo patients compared to controls [
101,
102].
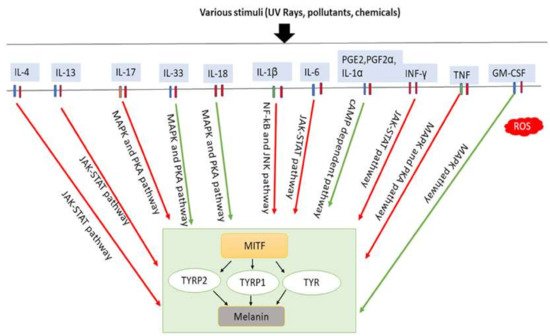
Figure 3. Involvement of inflammatory factors in melanogenesis. Inflammatory factors including IL-33, IL-18, and GM-CSF promote melanogenesis by stimulating the MAPK and PKA pathways either simultaneously or individually. In addition, PGE2, PGF2α, and IL-1α promote melanogenesis by stimulating cAMP-dependent pathway. In contrast, inflammatory factors such as IL-4, IL-13, IL-6, IL-17, IL-1β, TNF and IFN-γ inhibit melanogenesis by suppressing the PKA and MAPK, or JAK-STAT, or NF-κβ and JNK pathways. IL-33: interleukin-33; IL-18: interleukin-18; PGE2: prostaglandin E2; PGF2α: prostaglandin F2α; IL-1α: interleukin-1α; GM-CSF: granulocyte-macrophage colony stimulating factor; IL-4: interleukin-4; IL-13: interleukin-13; IL-6: interleukin-6; IL-17: interleukin-17; IL-1β: interleukin-1β; TNF: tumor necrosis factor; IFN-γ: interferons-γ; PKA: protein kinase A; MAPK: mitogen-activated protein kinase; JNK: c-Jun N-terminal kinases; JAK-STAT: Janus kinase-signal transducer and activator of transcription; NF-κβ: nuclear factor kappa-B; TYR: tyrosinase; TRRP1: tyrosinase-related protein-1; TRRP2: tyrosinase-related protein-2. Red arrows indicates inhibition; green arrows indicate promotion.
IL-13 is a typical Th2 cell cytokine also secreted by CD4 cells, natural killer T cells, mast cells, basophils, eosinophils, and group 2 innate lymphoid cells (nuocytes) [
103] sharing a receptor of similar structure and JAK2-STAT6 signaling pathway with IL-4 [
104,
105]. Recently, it has been found that activated human epidermal γδ T cells produce larger amounts of IL-13 than IL-4 [
106] and enhance IL-13 production following treatment with Ginsenoside F1 (GF1) extracted from plant genus Panax. This plays a role in the skin-whitening effect of GF1 via effective suppression of tyrosinase and DCT, indicating that IL-13 could be a direct modulator of melanogenesis through the JAK2-STAT6 signaling pathway in skin disorders accompanied by enhanced T-cell responses, such as atopic dermatitis and vitiligo [
107].
IL-17 is known to prevent bacterial and fungal infections [
108] and can induce the release of large amounts of proinflammatory cytokines from a variety of cells, such as epithelial cells, endothelial cells, and fibroblasts [
109], and its effects are vastly increased when cooperating with TNF [
110]. IL-17, a proinflammatory cytokine, is produced primarily by Th17 cells, as well as by other immune cells, including neutrophils, natural killer cells, mast cells, αβ and γδT cells, and group 3 innate lymphoid cells. Studies have revealed IL-17 can synergize with TNF to inhibit signaling pathway for melanogenesis, thereby inhibiting pigmentation [
93]. For example, studies on psoriasis lesions showed that overexpression of IL-17 and TNF increase melanocyte number and cause a simultaneous decrease in pigmentation signaling. Neutralization of TNF and/or IL-17 with mAbs revealed that both can recover growth and pigment production of melanocytes, which may contribute to pigmentation changes associated with psoriasis treatment [
93].
Table 1. Mechanisms of cytokines in the regulation of melanogenesis.
Recent research has shown that IL-33 can improve melanin biosynthesis in NHEM, stimulate phosphorylation of p38 and CREB, and increase TYR, TYRP1 and TYRP2 (DCT) expression through MITF, resulting in augmentation of melanogenesis [
112]. Abundant IL-33 mRNA is found in keratinocytes induced by UVB [
111] and fibroblasts [
120,
121], suggesting injury on skin causes IL-33 release, resulting in skin pigmentation. Initially, IL-33 can induce mast cells to produce proinflammatory cytokines and chemokines (including monocyte chemoattractant protein-1 and macrophage inflammatory protein-1α) [
122,
123,
124,
125,
126], leading to activation of macrophages [
127,
128,
129], CD 4+T cells, basophils, dendritic cells and neutrophils [
122,
129,
130,
131,
132], and likely promoting Th2-skewed skin inflammation, which is another indirect effect on pigmentation by IL-33 [
133]. It has also been discovered that IL-33 activates and binds its receptor suppressing tumorigenicity 2 (ST2)L/IL-1RAcP on different types of cells, including mast cells and Th2 cells [
134]. Then, IL-33 activates NF-κB, suggesting that it regulates outcome of diseases such as atopic dermatitis, which can trigger pigmentation [
134].
IL-18 may also modulate both innate and adaptive immunity and its dysregulation can cause autoimmune or inflammatory disease, but some studies recently suggest its participation in pigmentation regulation. It has been observed that IL-18 promotes melanogenesis and upregulates TYRP1 and TYRP2 expression [
113,
118] by activating pathways and increasing cascade expressing MITF [
113,
118]. These results indicate the involvement of IL-18 in the regulation of pigmentation by regulating melanogenesis.
IL-1 is an important proinflammatory cytokine involved in initiating inflammation and immunological reactions [
135], which contributes to increased tumor invasiveness, metastasis, and angiogenesis under chronic inflammatory conditions [
115]. IL-1 is produced predominantly from monocytes and macrophages [
136], although it is also produced by another types of cells, such as keratinocytes and fibroblasts [
137]. IL-1 possesses two forms, IL-1α and IL-1β [
138]. Although they share only 24% identity in protein sequence, IL-1β and IL-1α fold in an extremely similar manner and bind to the same receptor—the type I and type II IL-1 receptor (IL-1RI, IL-1RII, respectively) [
139]. The expression patterns of IL-1α and IL-1β vary among cell types [
140]. IL-1α is predominantly produced by keratinocytes. Keratinocytes secrete both IL-1α and IL-1β molecules in vitro [
137], in addition to the expression of IL-1RI, IL-1RII [
141]. The signal transduction is initiated by binding to IL-1RI [
142] which can inhibit tyrosinase activity and melanogenesis [
15,
115] and also stimulates human fibroblasts to produce keratinocyte growth factor (KGF) [
143]. IL-1α released from keratinocytes has been shown to regulate the homeostasis of mammalian skin [
144] and sufficient to trigger skin inflammation in humans [
141,
144] and produce more IL-1α upon UVB exposure [
145]. It is documented that KGF-induced TYR expression in primary melanocytes [
114]. The combination of KGF and IL-1α increases melanin deposition and is responsible for initial stage of human solar lentigines [
114]. On the other hand, it was observed that the expression of MITF-M was inhibited after IL-1β treatment to a panel of melanoma cell lines. The inhibitory effects of IL-1β on melanogenesis were removed by inactivation of NF-κB and JNK pathways, suggesting IL-1β could inhibit melanogenesis by downregulating MITF-M through NF-kB and JNK pathways [
115].
IL-6 is produced by numerous different cell types including keratinocytes, fibroblasts, and dermal endothelial cells and plays a critical role in regulating acute phase response, hematopoiesis, inflammation, metabolic control, liver regeneration, bone metabolism and cancer progression by promoting cell growth, survival, and differentiation [
146]. It has been revealed in recent studies that the IL-6 cytokine elicits a dose-dependent decrease in tyrosinase activity, thus inhibiting melanocyte proliferation and melanogenesis [
15].
PGE2 is the most abundant prostaglandin released by keratinocytes in response to UVR and inflammatory conditions such as wound healing and stimulates the formation of dendrites in melanocytes [
147,
148]. A wide range of physiological processes in the skin, including immune function, carcinogenesis, cutaneous barrier function, cell growth, and differentiation are regulated by PGE2 [
149,
150,
151,
152]. Biosynthesis of PGE2 is controlled by phospholipase A2 (PLA2), cyclooxygenase (COX), and prostaglandin E synthase (PGES) enzymes. PGF2α is produced by fibroblasts and keratinocytes and stimulates melanocyte dendrite formation and tyrosinase activation, but not proliferation [
151]. PGE2 binds to four distinct G-protein coupled receptors (EP1–4) and EP4 receptor signaling stimulates cAMP production in melanocytes. EP3 receptor-lowered basal cAMP levels suggest relative levels of activity of these receptors’ control effects of PGE2 on cAMP in melanocytes that stimulates tyrosinase activity and proliferation [
116,
151].
IFN-γ is also a common secretory cytokine in the skin and the most important endogenous mediator of immunity and inflammation [
118]. Th1 lymphocytes, CD 8+ cytotoxic T lymphocytes and NK cells mainly release the proinflammatory cytokine IFN-γ [
153]. However, IFN-γ is also secreted by other cells, including antigen-presenting cells, B cells and NKT cells [
154,
155,
156]. A clear relationship between increased IFN-γ signaling and a decreased number of stage III and IV melanosomes in the lesional skin of leprosy patients have been shown [
117]. Specific IRF1 binding sites are in the promoter region of pigmentation-related genes such as TYR, TYRP2 (DCT). It is shown that IFN-γ negatively regulates the expression of DCT and other pigmentation-related genes, providing strong evidence that IFN-γ had a significant role in downregulating melanosome maturation [
117] —i.e., skin hypopigmentation [
117]. Two other crucial mediators of skin pigmentation, α-MSH and TGF-β, are involved in the protective tanning response and maintenance of melanocytes in an immature state [
157,
158] and are released during inflammation or UV exposure. Thus, Natarajan VT et al. proposed an αβγ-cytokine regulatory model of melanogenesis [
117]. Recent studies in various mouse models of vitiligo have demonstrated that local IFN-γ accumulation produced by melanocyte-specific CD 8+ T cells plays an important role in skin depigmented spots [
159,
160] and also revealed increased IFN-γ is essential for vitiligo pathogenesis by inducing apoptosis of melanocytes [
161]. Moreover, IFN-γ can upregulate STAT1 phosphorylation, and JAK1 inhibitors can suppress inhibiting effect of IFN-γ, thus contributing to melanogenesis. Other studies have also shown that IFN-γ inhibits IL-18-induced melanogenesis [
118].
TNF functions by binding to two different receptors, TNFR1/p55 and TNFR2/p75 [
92], and is secreted mainly by monocytes and macrophages, and also by keratinocytes, dendritic cells, Th1, Th17 and Th22. TNF induces inflammation through activation of vascular endothelial cells and immune cells and plays a crucial role as a regulator of lymphoid tissue development by controlling apoptosis [
92]. In several autoimmune diseases, increased levels of TNF have been found at inflammation sites, such as in the lesional skin of psoriasis patients [
162]. Overexpression of IL-17 and TNF synergistically modulate cytokine expression, increase melanocyte number while suppressing pigmentation signaling [
93]. Therapeutic neutralization of TNF and IL-17 with mAbs resulted in a rapid recovery of pigmentation-related gene expression in psoriasis lesions, indicating IL-17 and TNF can affect pigment production in melanocytes, which may contribute to the hypopigmentation associated with psoriasis. For example, after 24–48h treatment with both IL-17 and TNF of melanocytes, decreased levels of c-KIT, MC1R, MITF, and TYRP2 were observed [
93]. Consequently, the levels of tyrosinase and melanin were significantly reduced [
93]. It has been suggested the combination of IL-17 and TNF can inhibit melanogenesis through PKA and MAPK signaling pathways [
92,
93] and blocking TNF can lead to rapid restoration of pigmentation gene expression in psoriatic lesions, indicating that anti-TNF has potential for treating depigmented dermatosis [
93]. IL-33 expression is induced by IL-17 and IFN-γ in epidermal keratinocytes and induces melanogenesis, which creates a negative feedback loop in epidermal pigmentation [
163,
164].
GM-CSF may be a vital factor in the melanogenesis of the skin because of its role in promoting melanocyte proliferation and melanin synthesis. GM-CSF is produced by mononuclear macrophages, keratinocytes and Th cells [
119]. It has been found GM-CSF synthesis and secretion by HaCaT cells increased cell proliferation in the wound healing process and contributes to wound healing and melanogenesis at the site of injury [
165]. In addition, to predict the prognosis of transplantation of cultured autologous melanocytes (TCAM) in vitiligo patients, increased serum levels of GM-CSF could be used as the serum biomarkers [
166].