1. The antiphospholipid syndrome (APS)
The antiphospholipid syndrome (APS) is characterized by thrombosis and/or pregnancy morbidity with the persistent presence of antiphospholipid antibodies (aPLs). Laboratory criteria for the classification of APS include the detection of lupus anticoagulant (LAC), anti-cardiolipin (aCL) antibodies and anti-β2glycoprotein I (aβ2GPI) antibodies () [
1]. Both venous and arterial thrombosis can occur in APS patients, with venous thrombosis being slightly more prevalent. Ischemic stroke and transient ischemic attacks (TIAs) are the most common manifestations in the arterial circulation [
2]. aPLs have been described to interfere with endothelial cells and the complement system, which may also be involved in the pathogenesis of arterial thrombosis [
2].
Table 1. Updated antiphospholipid syndrome (APS) classification criteria. APS is determined in the presence of at least one of the clinical criteria and one of the laboratory criteria. Adapted from Miyakis et al. [
3].
The Von Willebrand Factor (VWF) VWF is a multimeric protein that circulates in a latent conformation, which can be converted into an active conformation. Active VWF is able to bind to the platelet-receptor glycoprotein-Iba (GPIba) and plays an important role in arterial thrombosis by mediating platelet adhesion and aggregation [
4]. In healthy individuals, only a small portion of VWF circulates in its active conformation [
5]. However, it has been shown that active VWF is increased in various pathological conditions, for example, Von Willebrand disease type 2B, congenital/acquired thrombotic thrombocytopenic purpura (TTP), HELLP-syndrome (hemolysis, elevated liver enzymes and low platelets), malaria and APS [
6].
β2GPI is known to bind VWF and to inhibit platelet adhesion under flow conditions [
4]. Moreover, aβ2GPI antibodies are able to inhibit the binding of VWF to β2GPI, thereby increasing the amount of active VWF present in the circulation. This indicates that β2GPI may act as a regulator of VWF-platelet interactions that are altered in the presence of aβ2GPI antibodies.
2. Von Willebrand Factor (VWF)
The VWF is a plasma glycoprotein of ca. 270 kDa (in the form of mature VWF) that is synthesized in megakaryocytes, platelets and endothelial cells (ECs). VWF plays a role in primary hemostasis and acts as an indicator for inflammation [
7,
8]. Another role is to bind factor (F) VIII, thereby protecting it from clearance, and to transport FVIII to the site of injury [
9]. VWF is mostly known due to its role in several severe diseases such as von Willebrand disease (VWD) [
10] and acquired von Willebrand syndrome (aVWS) [
11]. Quantitative and/or qualitative abnormality in the adhesive plasma protein VWF will lead to VWD, one of the most common inherited bleeding disorders. In a recent study, Simurda et al. utilized plasma VWF carrying little FVIII and successfully inhibited VWD type III [
12]. Thrombotic thrombocytopenic purpura (TTP) is a life-threatening disease due to a quantitative or qualitative defect in ADAMTS13 resulting in the increased presence of ULVWF, finally resulting in severe thrombotic events such as systemic platelet aggregation, organ ischemia [
13].
As a multi-domain glycoprotein, VWF glycosylation occurs in both ER and Golgi apparatus [
14]. Firstly, VWF co-translation folding occurs when entering the endoplasmic reticulum from the ribosome, while being accompanied by N-linked glycosylation mediated by oligosaccharyltransferase (OST) to promote correct protein folding. Secondly, N-linked and O-linked glycosylation presented in Golgi apparatus leads to the addition of sialylation, sulfation and blood group determinants to VWF multimers, affecting platelet adhesion, interaction with ADAMTS13 and VWF clearance [
15,
16,
17,
18].
The original product (pre-pro-VWF) coded by the
VWF gene encompasses a signal peptide (SP), a VWF propeptide (VWFpp) and a mature VWF. A complete VWF monomer includes four types of homologous domains, which, in order from N- to C-terminal, are: SP -D1 -D2 -D’ -D3 -A1 -A2 -A3 -D4 -C1 -C2 -C3 -C4 -C5 -C6 -CK. In the endoplasmic reticulum (ER), the signal peptide is cut from the pre-pro-VWF and dimerization of pro-VWF monomers is followed by disulfide bonds near C-terminals [
19,
20]. VWF multimers are generated via N-terminal disulfide bonds in the Golgi apparatus through D1 and D2 domains supported by the VWF propeptide, with the process of VWF propeptide removal under the catalysis of furin. This process can lead to the development of ultra-large VWF multimers that can range from 500 kDa to more than 20,000 kDa [
19,
21]. After these biomodifications are done in the ER and Golgi apparatus, the mature VWF (VWF antigen (VWF: Ag)) and VWFpp are partly secreted into the plasma. However, most of them will be stored in the ultra-large (UL) form within the Weibel–Palade bodies of ECs or the α-granules of megakaryocytes and platelets [
20,
22,
23]. Acute endothelial cell activation can be observed by measuring both VWFpp and VWF: Ag, knowing that the half-life of VWFpp is shorter [
20].
Under normal conditions, VWF circulates in the plasma in a spherical conformation [
24]. Upon vascular damage, VWF is released by ECs and binds via its A1 domain (to collagen type I, III, IV and VI) and A3 domain (to type I and III) which is present in the perivascular connective tissue of the damaged vessel wall [
20,
21,
25,
26,
27]. The binding of VWF to collagen will induce a conformational change that results in a stretched conformation thereby exposing the A1 domain [
28]. The VWF-A1 domain can interact with platelet receptor GPIbα [
29], allowing adhesion of circulating platelets [
30,
31,
32]. Moreover, UL-VWF is also released into the plasma from the ECs and platelets and will likewise participate in platelet adhesion and aggregation. The excessive presence of UL-VWF multimers may result in the development of thrombosis as, due to its larger size, UL-VWF has more binding sites for platelets and collagen when it is unfolded [
33]. However, in normal conditions, the development of thrombosis is prevented by ADAMTS-13 (a disintegrin and metalloprotease), which is responsible for degrading UL-VWF into much smaller proteins via the cleavage site on the A2 domain of VWF [
34].
In atherosclerotic lesions, VWF can act as a bridge between collagen and platelets, as well as between platelets themselves, thereby being a major contributor to the development of arterial thrombosis. A meta-analysis investigating the relationship between the levels of VWF and ADAMTS-13 with arterial thrombosis illustrated that high levels of VWF were associated with coronary heart disease and ischemic stroke, while ADAMTS-13 levels were lower in stroke patients than in controls and patients with coronary heart disease [
35]. Pickens et al. used an
ADAMTS-13 gene knock-out mouse model to verify the importance of ADAMTS-13 in arterial thrombus formation. The authors found that the thrombus formation velocity was significantly lower in transgenic mice that expressed recombinant human ADAMTS-13 compared to ADAMTS-13 knock-out mice but was similar to wild-type mice [
36]. In accordance, Masias et al. showed a lower activity of ADAMTS-13 in patients with ischemic stroke and myocardial infarction, supporting the finding that ADAMTS-13 is involved in arterial thrombosis [
37]. Moreover, Verhenne et al. hypothesized that the interaction between GPIbα and VWF is involved in the development of stroke [
38]. Brait et al. found that a CD69 deficiency can promote thrombophilia by increasing the circulating plasma levels and activity of VWF [
39]. In addition, in vivo experiments with DTRI-031, a novel aptamer designed by Nimjee et al. that inhibits VWF-mediated platelet adhesion, could inhibit platelet aggregation and thrombosis in mice [
40].
Furthermore, the incidence of arterial thrombosis is significantly reduced in patients with VWD and there is increasingly more evidence that inflammation can cause VWF-mediated thrombosis [
41]. Possible mechanisms could be the activation of the endothelium, secretion of VWF, assembly of hyper-adhesive VWF strings and fibers, reduced cleavage by ADAMTS13, as well as adhesion and deposition of VWF-platelet-thrombi in the vasculature [
42].
4. Platelet Activation
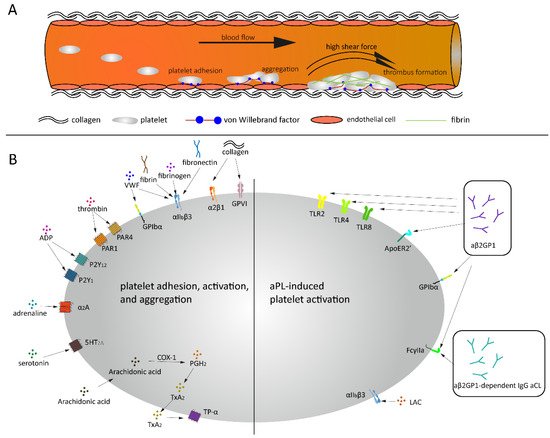
Thrombus formation is a process that consists of two interdependent mechanisms that involve the activation of platelets and activation of the coagulation system. When a vessel wall is damaged, the collagen that is located in the extracellular matrix (ECM) will be exposed to the blood. As mentioned previously, VWF exposes its A1 domain under high shear or bound to collagen A. Subsequently, this A1 domain binds to the GPIbα receptor on platelets, which will make the platelets roll over the collagen [
29]. This rolling creates the ideal condition for platelet glycoprotein VI (GPVI) and integrin α2β1 to bind to the subendothelial collagen matrix tightly. Platelets adhering to the exposed collagen will become activated, change in shape, secrete their granule content and form an aggregate with other platelets. Additionally, platelets will recruit VWF [
47], fibrinogen [
48], fibrin and/or fibronectin [
49] via integrin αIIbβ3, which in turn will also mediate platelet adhesion and activation [
50,
51]. Interestingly, the binding between platelet GPIbα and VWF is flexible and allows the platelet to translocate over VWF, while the binding between platelets via integrin αIIbβ3 with VWF and fibrinogen is more rigid [
48].
Figure 1. Thrombus formation under high shear and aPL related platelet activation. (A) Platelets bind to endothelial cells and collagen via VWF under high shear force, followed by platelets rolling over collagen and thrombus formation. (B) Under normal conditions, a variety of agonists and ligands bind their corresponding receptors, inducing platelet adhesion, activation and aggregation. aβ2GPI and aCL antibodies induce platelet activation in APS patients.
The platelet plug formed can be consolidated via clot retraction which is mediated via the binding of αIIbβ3 on actin filaments [
52].
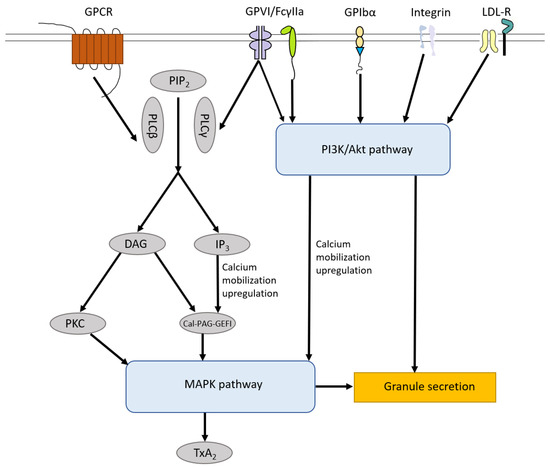
Activated platelets release soluble agonists, for example, adenosine diphosphate (ADP), Thromboxane A2 (TXA2) and thrombin, which induce autocrine and paracrine platelet activation. A wide variety of receptors are expressed on the platelet surface, for example, G protein-coupled receptors (GPCRs), glycoprotein receptors, CLEC-2, Toll-like receptor 2 (TLR2) and platelet endothelial aggregation receptor 1 (PEAR1). Most of them are transmembrane receptors that are responsible for clot formation due to trauma or bleeding but also for signaling transmission and other physiological processes, for example, inflammatory reactions. These receptors also play an important role in pathological conditions as tumor metastasis and autoimmune diseases. The most abundant receptor present on the platelet surface is GPCR, which is a family consisting of seven-transmembrane (7TM) α-helices and a G protein [
53,
54,
55]. To date, about 1000 unique GPCRs have been identified in the human genome, which participate in extracellular ligand recognition and intracellular signaling transduction [
56]. GPCRs are expressed in hematopoietic and vascular tissues and mediate platelet activation, such as protease-activating receptors (PAR family), ADP receptors (P2Y1 and P2Y12), TXA2 receptors (TP-α), prostaglandin E2 receptors (EP family), adrenoreceptor (α2A) and serotonin receptor (5HT2A), as well as platelet inhibition, such as prostacyclin receptor (IP), adenosine receptor (AR) and prostaglandin D2 receptor (DP1 receptor) (B) [
57]. When the receptors that activate the platelets are stimulated, the content of the platelet granules is released, which in turn will lead to more platelets adhering, spreading and secreting their granule content. The propagation of the clot formation is ensured by the two pathways: the inside-out signaling pathway (i.e., when activated αIIbβ3 binds to its ligands, e.g., VWF and fibrinogen) and the outside-in signaling pathway (i.e., a series of intracellular signaling events that start after the platelet is activated) [
58]. Sokol et al. explored 12 selected single nucleotide polymorphisms (SNPs) within seven specific genes in patients suffering from sticky platelets syndrome (SPS). They found that platelet endothelial aggregation receptor 1 (PEAR1) is associated with increased overall platelet aggregation and reduced responsiveness to aspirin and may act as a protective factor for DVT in patients with SPS type II which is stimulated by epinephrine [
59].