RANK ligand (RANKL) is a member of the tumor necrosis factor alpha superfamily of cytokines. It is the only known ligand binding to a membrane receptor named receptor activator of nuclear factor-kappa B (RANK), thereby triggering recruitment of tumor necrosis factor (TNF) receptor associated factor (TRAF) adaptor proteins and activation of downstream pathways. RANK/RANKL signaling is controlled by a decoy receptor called osteoprotegerin (OPG), but also has additional more complex levels of regulation.
- RANK
- RANKL
- immunotherapy
- checkpoint inhibition
- cervical cancer
- microenvironment
1. Introduction
Although cervical cancer (CC) is traditionally regarded as a preventable disease, it continues to be a major health problem in many parts of the world. Globally it is the fourth most common cancer in women, with most cases diagnosed in an advanced stage [1]. A chronical infection with a human papilloma virus (HPV) plays a major etiological role in about 95% of invasive CCs [2]. Before developing an invasive carcinoma, cervical cells go through different precancerous stages, called cervical intraepithelial neoplasias (CINs). These are classified according to their grade of dysplasia: very mild to mild (CIN I), moderate (CIN II), and severe dysplasia to carcinoma-in-situ (CIN III). Most mild dysplastic lesions disappear spontaneously, but a significant proportion of the CIN III lesions may progress into invasive CC if left untreated [3]. These precancerous lesions have a likelihood of about 95% to be picked up in by screening programs using cytology and/or HPV testing on cervical smears [4]. They can be effectively eradicated by minimally invasive excision or ablation techniques, truly preventing CC. Primary prevention of CC is possible by vaccination against HPV. Patients diagnosed with early invasive CC are preferentially treated surgically: by a cone biopsy, hysterectomy, or radical hysterectomy with pelvic lymphadenectomy or sentinel node biopsy depending on their stage [5,6]. Curative concurrent chemotherapy combined with radiotherapy is the first treatment of choice for patients with more advanced CC (Federation International de Gynecologie et Obstetrie (FIGO) stages IB2 to IVA), but can also be used in earlier stages depending on comorbidity and patient preference [6]. Over the last decades metastatic disease was treated with platinum-based chemotherapy, but there has been little improvement in the efficacy of the systemic treatment [3]. The short-lived responses to chemotherapy seen in patients with advanced cervical cancer indicate that the disease is relatively chemorefractory and up to now most new targeted drugs provided no extra survival benefit [3,4]. The Gynecologic Oncology Group (GOG) 240 phase III trial showed that addition of bevacizumab to the current golden standard (combination of topotecan-paclitaxel or cisplatin-paclitaxel) in this population is associated with a moderate increased overall survival (16.8 months vs. 13.3 months) compared to the same cytostatic combinations without antiangiogenic medication. At 36 months the likelihood to be alive was about 10% in both patient groups illustrating the dismal prognosis of these patients [7]. Inoperable CC is likely to remain highly prevalent during the next decades as screening programs and vaccination campaigns against human papilloma virus (HPV) have suboptimal participation rates, are not completely effective, and are still unavailable in most countries [4].
It has been known for decades that HPV infection plays a crucial role in triggering most CCs, but only recently the regulatory networks involved have been unraveled [2]. Important driver pathways in CC carcinogenesis are associated with avian myelocytomatosis viral oncogne holog (MYC) signaling, cell cycle deregulation, TGFβ-signaling, mitogen-activated protein kinase (MAPK) signaling, and chromatin modeling [8]. In 2017, a comprehensive landmark study on invasive CC was conducted as part of The Cancer Genome Atlas (TCGA) project, aiming to identify relevant molecular events that drive tumorigenesis [9]. Integrated clustering identified keratin-low squamous, keratin-high squamous, and adenocarcinoma-rich clusters defined by different HPV and molecular features. Higher activation of p53, p63, p73, activator protein 1 (AP-1), MYC, hypoxia-inducible factor 1-alpha (HIF1A), fibroblast growth factor receptor 3 (FGFR3), and MAPK pathways were found to be distinguishing features of squamous cell carcinomas, while adenocarcinomas exhibited higher inferred activation of estrogen receptor alpha (ERα), forkhead box protein A1 (FOXA1), FOXA2, and FGFR1 signaling [9]. The authors could identify new frequently mutated genes (transforming growth factor beta 2 (TGFBR2), epiderma growth factor receptor 3 ERBB3, cysteine-aspartic acid protease 8 CASP8, SHKBP1 binding protein 1, and human leucocyte antigen A HLA-A, confirm previously reported significant gene mutations (phosphatidylinositol-4,5biphosphate 3 kinase PIK3CA, KirstenRat sarcoma virus KRAS, phosphatase and tensin homolog PTEN, MAPK1, nuclear factor erythroid 2 like 2 NFE2L2, AT-rich intercative domain-containing protein 1A ARID1A, EP300, F-box/WD repeat-containg protein 7 FBXW7, HLA-B), and detected striking apolipoprotein B mRNA editing enzyme catalytic polypeptide-like APOBEC mutagenesis patterns. They also discovered amplifications in the well-known checkpoint controlling immune targets programmed death ligand 1 (PD-L1, CD274) and PD-L2 (PDCD1LG2) in the keratin-low and -high squamous cancers, but not in the adenocarcinoma enriched group. In addition, amplifications in the long non-coding RNA of the breast cancer anti-estrogen resistance 4 (BCAR4) gene, which regulates the expression of perforin and granzyme A, were found [9]. This suggests that immunotherapeutic strategies may be active in a subset of cervical cancers. Preliminary studies confirm that responses are seen in a minority of patients with CC when they are treated with checkpoint inhibitors alone or in combination with chemo- or radiotherapy [10]. Recently, it has been suggested that RANK/RANKL inhibition has the potential to make tumors the more susceptible to checkpoint inhibition [11]. In the present paper, the literature (PubMed, Web of Science) on RANK/RANKL signaling in cervical cancer is reviewed up to March 2019. Furthermore, a translational window of opportunity study (DICER: denosumab in cervical cancer) starting in January 2019 is proposed investigating the effects of RANKL inhibition on the immune environment in patients with squamous carcinoma of the cervix.
2. The RANK/RANKL Signaling Network
The tumor necrosis factor (TNF) superfamily (TNFSF) is a superfamily of type II transmembrane proteins commonly containing the TNF homology domain [12]. This superfamily consists of more than 20 protein members, which can be released from the cell membrane by proteolytic cleavage by specific metalloproteinases to generate soluble cytokines [13]. The members of the TNFSF have in common that they interact with their cognate TNF receptor superfamily (TNFRSF) members [12,14]. Conserved residues provide specific inter subunit contacts. There is substantial crosstalk among related ligand receptor pairs and cytokines, and recent evidence suggests that signals can be generated from the receptor part (forward signaling) as well from the ligand part (reverse signaling) [12]. The TNFSF/TNFRSF axes are involved in regulating diverse biological processes, including embryogenesis, differentiation, proliferation, apoptosis, inflammation, and immune responses [14,15]. They provide a communication network that is essential for coordinating multiple cell types into an effective host defense system against pathogens and malignant cells [13].
RANKL is a type II homotrimeric transmembrane protein bearing close homology to TNFSF members TNF-related apoptosis inducing ligand (TRAIL), Fas ligand (FasL, TNFSF6), and TNF-alpha [15]. It has three known isoforms that are expressed as membrane bound (RANKL1, RANKL2) or as soluble secreted proteins (RANKL3) by cleavage of the membranous forms by the metalloprotease–disintegrin TNF-alpha convertase (TACE), or by alternative splicing [15,16]. The RANKL is encoded by the TNFS11 gene in humans and is also named osteoprotegerin ligand (OPGL), TNF-related activation induced cytokine (TRANCE), or osteoclast differentiation factor (ODF) [16,17]. It is the only known ligand binding to a membrane receptor named receptor activator of nuclear factor-kappa B (RANK (TNFRSF11A) [17,18]. Binding between RANKL and RANK induces trimerization of the receptor, which triggers recruitment of TNF receptor associated factors (TRAF), adaptor proteins, and activation of downstream signaling pathways (such as NF-κB, RAC-alpha serine/threonine-proteine kinase (AKT), protein kinase B (PKB), c-Jun N-terminal kinase (JNK), and the MAPK cascade) [19]. RANK/RANKL signaling is controlled by a decoy receptor called osteoprotegerin (OPG, TBFRSF11B), which interacts with RANK and TRAIL (Figure 1). OPG is a soluble glycoprotein that can exist either as a 60 kDa monomer or as a 120 kDa dimer but has no transmembrane or cytoplasmatic domains. The dimerization of OPG increases the affinity of OPG to RANKL dramatically [20]. OPG expression can be upregulated by several factors such as TRAIL, Wnt/β catenin signaling, IL-1β, TNFα, and estrogen, and down regulated by TGF-β and parathyroid hormone (PTH) [17,21]. The final inhibitory effect of OPG on RANKL is dependent on its binding to these ligands [22,23]. Recently a new receptor for RANKL, LGR4 (leucine-rich repeat-containing G-protein coupled receptor 4), has been identified, which suppresses canonical RANK signaling by competing with RANK to bind RANKL [24]. Data are emerging that complex additional levels of regulation of the RANK/RANKL network exist [17,25,26].
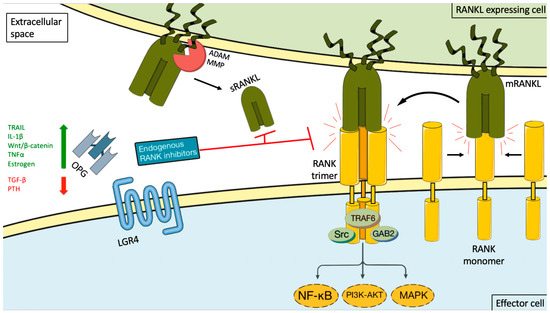
Figure 1. RANKL/RANK signaling and its endogenous inhibition. Binding between RANKL and RANK induces receptor trimerization, which triggers recruitment of TNF receptor associated (TRAF) adaptor proteins and activation of downstream signaling pathways (such as NF-κB, PI3K-AKT, and the MAP kinase cascade). This activity can be induced by either membrane-bound RANKL (mRANKL) or soluble RANKL (sRANKL). sRANKL is derived from the membrane-bound form through alternative splicing or proteolytic cleavage (for example, by matrix metalloproteinase (MMP) or disintegrin and metalloproteinase (ADAM) family members. RANK itself lacks kinase activity, and its signaling is initially mediated by adaptor molecules, such as TRAF proteins (including TRAF6), GRB-associated-binding protein 2 (GAB2), and sarcoma proto-oncogene tyrosine-kinase (SRC). The signaling cascade is controlled by a decoy receptor called osteoprotegerin (OPG) and leucine-rich repeat-containing G protein-coupled receptor 4 (LGR4). OPG expression can be upregulated by several factors such as TRAIL, IL-1β, Wnt/β catenin signaling, TNFα, and estrogen, and down regulated by TGF-β and PTH. The endogenous inhibitors bind both the soluble and membrane-bound RANKL forms, thereby preventing it from interacting with RANK. The green arrow stand for upregulation, the red arrow for inhibition and the black arrows for possible interactions and the curved black arrow for the result of interaction.
3. RANK-Mediated Signaling and Cervical Cancer
Until recently little was known on the role of the RANKL-RANK signaling network in cervical cancer. In 2015, Shang et al. showed that RANK and RANKL were co-expressed in carcinoma of the uterine cervix. They assessed the expression of RANKL and RANK in the cervical cancer cell lines SiHa and HeLa and in twelve cervical cancer tissues. Stronger membrane staining for RANKL and RANK was detected in the cell lines and in cancer cells compared to precancerous cells [65]. Other groups confirmed that cytoplasmatic immunoreactivity for RANKL is weak in normal cervical tissues and became significantly higher during neoplastic transition from cervical intraepithelial neoplasia (CIN) I to CIN III and invasive CC [66,67]. In a series of 110 patients with invasive CC, the expression of RANK mRNA was significantly correlated with tumor pathological grade, clinical stage, depth of invasion, and lymph node metastasis. RANK is a direct binding target of KLF5 (Krüppel-like factor 5), a zinc finger containing protein [67]. In vitro experiments showed that KLF5 promotes proliferation, migration, and invasion of HeLa cells by up-regulating the transcription of RANK. Tumor necrosis factor (TNF)-α induced KLF5 expression by activating the p38 signaling pathway [67]. High KLF5 or RANK expression increased the risk of death in patients with squamous CC [67]. It was previously demonstrated that hypoxia can upregulate the expression of RANK and RANKL, and increases RANKL-induced cell migration via the PI3K/AKT-hypoxia inducible factor-1α (HIF-1α) pathway [68]. Therefore, these inflammatory factors and local hypoxia may contribute to a high level of RANKL/RANK in cervical cancer cells.
Cervical cancer cells secrete soluble RANKL (soRANKL) [66]. Blocking RANKL/RANK interaction by recombinant OPG or anti-RANKL neutralizing antibodies significantly decreases proliferation and increases apoptosis, suggesting that the stimulatory effect of RANKL on the growth of cervical cancer cells may be mainly dependent on membrane RANKL/RANK interaction [66]. In an immunodeficient mouse model, RANK-knockdown suppresses the metastatic potential of Hela cells. Antibodies against RANKL reduce the secretion of IL-8 by SiHa and Hela cells, thereby inducing apoptosis and reducing proliferation. On the other hand, recombinant IL 8 has the opposite effect [66]. This indicates that RANKL may stimulate cervical cancer growth by activation of the IL-8 pathway. Jia et al. recently showed that the expression of IL-8 was significantly higher in cervical carcinoma samples compared to normal controls and that migration and proliferation of Hela cells increased after treatment with IL-8 [69]. Survival of cervical cancer patients with high intratumoral IL-8 expression is significantly worse compared to patients with low IL-8 in their tumors [70]. Strikingly, these authors identified colocalization between IL8 and CD66 (tumor associated macrophage staining), and a correlation with microvessel density counts, vascular endothelial growth factor VEGF, fibroblast growth factor FGF, and platelet derived growth factor PDGF levels. This suggests that IL-8 is likely to be involved in angiogenic growth and creating an immune suppressive environment. In a xenograft mouse model using cervical cancer cells with a high IL-8 production (CaSki cells) treatment with an antibody against IL-8 resulted reduced tumor growth and lymph node metastases and prolonged survival significantly [71].
Only a minority of genital human papillomavirus (HPV) infections progress into (pre)neoplastic lesions [2]. Human papilloma virus can use multiple mechanisms to evade the immune surveillance, to alter cell cycle control, and to facilitate the accumulation of DNA damage and malignant transformation of the cervical keratinocytes [36]. In an elegant set of experiments, Demoulin et al. identified RANK/RANKL signaling as a likely candidate driving the tolerogenic alterations in DCs in the genital (pre)neoplastic microenvironment [66]. They showed that squamous cervical and vulvar cancer cells (CVCC) secrete factor(s) that can affect the maturation and function of dendritic cells (DC) via RANK pathway signaling. Dendritic cells incubated with recombinant RANKL had reduced maturation characteristics, lower IL-12 and higher IL-10 expression, suggestive for an immunosuppressive status. OPG can inhibit this effect and restore DC functions. Dendritic cells cocultured with CVCCs induce FoxP3+ suppressive Treg cells from naive CD4+ T cells. Therefore, RANKL inhibition seems to be an attractive approach to increase the immunogenicity of cervical cancer. Garcia Paz et al. showed in a HPV16-positive tumor bearing syngeneic mouse model that treatment with the IL-12 gene increased the intratumoral expression of IL-12, IL-2, and gamma-interferon (being immune stimulatory) and reduced the expression of TGF-β1, IL4, and IL10 (known to be immunosuppressive) [72]. IL-12 gene transfer activated the immune response, inhibited tumor growth and prolonged survival of the mice. This proves the potential therapeutic concept of altering the immune response in cervical cancer.
Weak immunostaining for OPG could be detected in normal and metaplastic exocervix, while OPG expression was significantly higher in (pre)cancerous lesions regardless of their grade [73]. In a series of 218 patients with CC treated with primary surgery, 42% of patients with squamous carcinomas, and 49% of patients with adenocarcinomas had high OPG expression levels. High OPG expression was correlated with high FIGO stage, tumor grade, presence of lymph node metastases, and poor overall survival. It proved to be an independent prognostic factor in multivariate analysis [73]. Osteoprotegerin was amongst six proteins having significantly different levels in CC patients compared to controls and CIN. This confirms that OPG may be involved in cervical cancer carcinogenesis and has the potential to be used as a biomarker [74].
In an in silico study comparing stromal and immune response related genes in normal cervix versus cervical carcinoma (unpublished data), we identified TMPRSS11D (encoding a serine-like trypsine protease) and CLCA2 (human chloride channel accessory-2 gene) among the top ten differentially expressed genes [8]. TMPRSS11D directly interacts with OPG [75]. In mice the ion channel CLCA2 is significantly induced by RANKL stimulation [76]. CLCA2 encodes a member of the calcium-activated chloride channel regulator (CLCR) family of proteins that regulates the transport of chloride across the plasma membrane. Expression of CLCA2 is upregulated by the tumor suppressor protein p53 in response to DNA damage and this has a significant effect on cell migration and invasion [77].
The above data indicate that RANK/RANKL signaling is an important player involved in the initiation, progression, proliferation, migration, and invasion of CC, and in modulating the immune environment. Theoretically RANKL inhibition is an intriguing target for CC for two main reasons. First, the high levels of expression of RANK and its ligand in a large proportion of CC’s, the in vitro effects of RANKL inhibition on proliferation and the correlation with prognosis of high RANKL expression make it likely that there may be a direct antiproliferative effect on the tumor cells and a reduction of the metastatic potential. Secondly there is abundant circumstantial evidence suggesting that the effects on the microenvironment may (partially) reverse an immunosuppressive status [78]. As CC is a chronic inflammatory disease with a high mutational load combining RANKL inhibition with immunotherapy is an attractive therapeutic option which should be further explored [79].
This entry is adapted from the peer-reviewed paper 10.3390/ijms20092183