Administration of the chemotherapeutic agent cisplatin leads to acute kidney injury (AKI). Cisplatin-induced AKI (CIAKI) has a complex pathophysiological map, which has been linked to cellular uptake and efflux, apoptosis, vascular injury, oxidative and endoplasmic reticulum stress, and inflammation. Despite research efforts, pharmaceutical interventions, and clinical trials spanning over several decades, a consistent and stable pharmacological treatment option to reduce AKI in patients receiving cisplatin remains unavailable. This has been predominately linked to the incomplete understanding of CIAKI pathophysiology and molecular mechanisms involved.
- cisplatin
- acute kidney injury
- nephrotoxicity
1. Introduction
1.1. Cisplatin
Cisplatin (cis-diamminedichloroplatinum II) is a platinum-containing antineoplastic drug first approved for clinical use in 1978 [1]. It is used extensively to treat a repertoire of malignancies per se or as a tailored combination in treatment [1]. Cisplatin is used to treat breast [2], cervical [2], oesophageal [3], bladder [4], small cell lung [5], and testicular cancers [6]. Cisplatin is also used as a combination therapy to treat high grade cancers such as osteosarcoma [7] and soft-tissue cancers including squamous cell carcinoma [8]. Cisplatin is one of the most potent and effective chemotherapies used to date [9], and its antitumor effects are well established [2][9][10]. However, the exact mechanism of cisplatin-induced cell death remains largely unknown. It is widely accepted that cisplatin causes 1–2 intrastrand or 1–3 interstrand crosslinks with purine bases on the deoxyribonucleic acid (DNA) strand [9][11]. This crosslinking impairs DNA repair mechanisms, inhibiting the production of a viable DNA replication template, stimulating cell-cycle arrest leading to cell death [12]. Irrespective of its potent anticancer properties and efficacy, the clinical usage of cisplatin is limited due to the severity of adverse side effects including ototoxicity and neurotoxicity [13][14] and its dose-limiting factor nephrotoxicity [12][15][16][17][18][19][20][21].
1.2. Nephrotoxicity
Nephrotoxicity results from a rapid decline of excretory mechanisms within the kidney [22], enhancing the accretion of waste products produced by protein metabolism (including urea, nitrogen, and creatinine) [22][23][24]. Acute kidney injury (AKI) is commonly caused by nephrotoxic injury to kidney tissue, resulting in acute tubular necrosis [25]. It can also result from inadequate urinal drainage [26]. Decreased drainage causes an increase in intratubular pressure and decreases glomerular filtration rate (GFR). Decreased GFR can additionally be stimulated by afferent arteriole vasoconstriction [27]. Despite improved prognosis following the removal of diuretics to promote volume expansion and hydration, the prevalence of cisplatin-induced AKI (CIAKI) remains high [28]. Although cisplatin-induced nephrotoxicity can manifest in a variety of ways, acute tubular necrosis (ATN) is the most prevalent [29]. In the clinical setting, AKI frequently occurs despite low-dose cisplatin administration [30]. The uptake of cisplatin into proximal tubular epithelial cells (PTEC) is the initiator of the toxic effects of cisplatin [31]. To date, despite burgeoned research, there is no intervention that adequately treats or prevents CIAKI in cancer patients [32]. Therefore, further understanding the molecular pathways and their interactions is essential in finding or developing a suitable pharmacological treatment to be used in conjunction with cisplatin.
1.3. Pathophysiology of Cisplatin-Induced AKI
A variety of molecular pathways and mechanisms have been investigated to determine the unknown pathological events caused by CIAKI. The key molecular mechanisms involved in cisplatin-induced nephrotoxic adverse effects include cellular uptake and accumulation, inflammation, oxidative stress, vascular injury, endoplasmic reticulum (ER) stress, and necrosis and apoptosis (Figure 1). A plethora of pharmacological agents (Table 1) and genetic alterations (Table 2) have been investigated in experimental preclinical studies of CIAKI. Despite the prevalence of nephrotoxicity in cisplatin-treated patients, its clinical application must be accompanied by other treatments to counteract its harmful effects while allowing it to exert its potent anticancer properties. Cisplatin cellular uptake is the initiator of the nephrotoxic effects, with several studies investigating the various therapeutic options that promote renoprotection (Figure 2). The purpose of this review is to collectively present the magnitude of preclinical studies in addition to presenting the clinical studies recently completed and currently being conducted for the treatment of CIAKI. The data from these studies illustrates the broad pathophysiological mechanisms involved and the potential for their inter-relationships. This review sheds light on the current failure in preclinic to clinic translatability given the lack of studies currently moving from animal models to human clinical trials. Despite the frequent protective therapies evaluated in models of CIAKI, there is no evidence of treatment progression with almost all therapies evaluated, posing a highly concerning issue for cisplatin patients.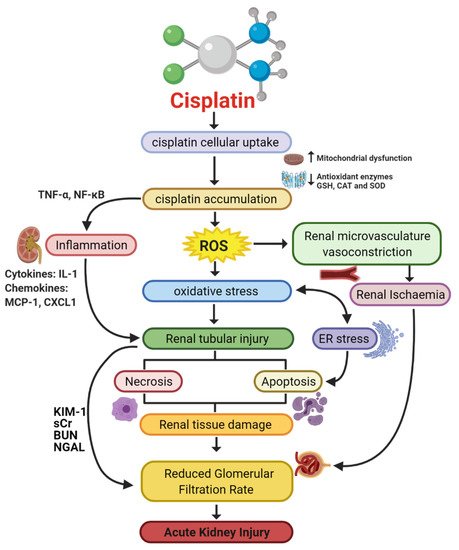 Figure 1. Pathophysiological map of the key molecular pathways demonstrated to play a role in the pathogenesis of cisplatin-induced acute kidney injury (AKI). The mechanisms associated with cisplatin-induced AKI (CIAKI) are complex, and the relationship between the key pathways remains unknown. However, it is believed that the detrimental nephrotoxic effect of cisplatin in renal tissue is due to platinum accumulation. Cisplatin accumulation triggers increased production of tumor necrosis factor alpha (TNF-α) [33][34] and reactive oxygen species (ROS), stimulating inflammation [35], oxidative stress [36], vascular injury [31], and apoptotic pathways [37]. The apoptotic mechanisms then promote renal tissue damage leading to the key clinical manifestation of nephrotoxicity (a reduction in glomerular filtration rate (GFR)) resulting in CIAKI. Abbreviations: GSH, glutathione; CAT, catalase; SOD, superoxide dismutase; TNF-α, tumor necrosis factor alpha; ROS, reactive oxygen species; ER stress, endoplasmic reticulum stress; and GFR, glomerular filtration rate. IL-1, Interleukin 1; MCP-1, monocyte chemoattractant protein 1; CXCL1, C-X-C Motif Chemokine Ligand 1; KIM-1, Kidney Injury Molecule 1; sCr, Serum Creatinine; BUN, Blood Urea Nitrogen; NGAL, Neutrophil gelatinase-associated lipocalin. Figure adapted from “Cisplatin nephrotoxicity: mechanisms and renoprotective strategies” by N. Pabla and Z. Dong, 2008, Kidney International, Volume 73, P994-1007, Copyright [2008] by the Elsevier.
Figure 1. Pathophysiological map of the key molecular pathways demonstrated to play a role in the pathogenesis of cisplatin-induced acute kidney injury (AKI). The mechanisms associated with cisplatin-induced AKI (CIAKI) are complex, and the relationship between the key pathways remains unknown. However, it is believed that the detrimental nephrotoxic effect of cisplatin in renal tissue is due to platinum accumulation. Cisplatin accumulation triggers increased production of tumor necrosis factor alpha (TNF-α) [33][34] and reactive oxygen species (ROS), stimulating inflammation [35], oxidative stress [36], vascular injury [31], and apoptotic pathways [37]. The apoptotic mechanisms then promote renal tissue damage leading to the key clinical manifestation of nephrotoxicity (a reduction in glomerular filtration rate (GFR)) resulting in CIAKI. Abbreviations: GSH, glutathione; CAT, catalase; SOD, superoxide dismutase; TNF-α, tumor necrosis factor alpha; ROS, reactive oxygen species; ER stress, endoplasmic reticulum stress; and GFR, glomerular filtration rate. IL-1, Interleukin 1; MCP-1, monocyte chemoattractant protein 1; CXCL1, C-X-C Motif Chemokine Ligand 1; KIM-1, Kidney Injury Molecule 1; sCr, Serum Creatinine; BUN, Blood Urea Nitrogen; NGAL, Neutrophil gelatinase-associated lipocalin. Figure adapted from “Cisplatin nephrotoxicity: mechanisms and renoprotective strategies” by N. Pabla and Z. Dong, 2008, Kidney International, Volume 73, P994-1007, Copyright [2008] by the Elsevier.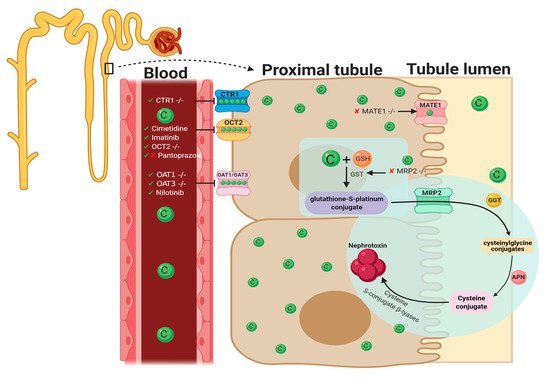 Figure 2. Graphical representation of key molecules and pathways involved in cisplatin transportation initiating nephrotoxic effects. Key transporters responsible for cellular uptake of cisplatin from the blood into PTECs resulting in a much greater platinum concentration compared to the blood. The key interventions trialed to date and their effectiveness in targeting CIAKI are also illustrated. Diagram details the cellular processes involved in the cellular uptake [38][39][40][41][42][43][44][45], efflux [46][47], and metabolism of cisplatin into a highly reactive thiol (nephrotoxin) [48] and the treatment targeted to prevent them.Table 1. Pharmacological interventions assessed for renoprotective effects against cisplatin-induced AKI in vitro and in vivo, papers published in 2020.
Figure 2. Graphical representation of key molecules and pathways involved in cisplatin transportation initiating nephrotoxic effects. Key transporters responsible for cellular uptake of cisplatin from the blood into PTECs resulting in a much greater platinum concentration compared to the blood. The key interventions trialed to date and their effectiveness in targeting CIAKI are also illustrated. Diagram details the cellular processes involved in the cellular uptake [38][39][40][41][42][43][44][45], efflux [46][47], and metabolism of cisplatin into a highly reactive thiol (nephrotoxin) [48] and the treatment targeted to prevent them.Table 1. Pharmacological interventions assessed for renoprotective effects against cisplatin-induced AKI in vitro and in vivo, papers published in 2020.
| Drug | Mechanism of Action | Findings | In Vitro | In Vivo | Reference |
|---|---|---|---|---|---|
| Aucubin | Anti-inflammatory | ↓ Markers of oxidative stress (HO-1 and 4-HNE) ↓ Apoptosis (caspase-3, caspase-9 and PARP) |
– | BALB/c mice | [38] |
| Curcumin | Anti-inflammatory, Antioxidative, oxygen-free radical scavenging, antifibrotic, and anticancer activities | ↓ Tubular Injury ↓ BUN ↓ sCr (rats) |
– | C57BL/6J mice/rats | [39][40] |
| Dexmedetomidine | Antiapoptotic via α2AR/PI3K/AKT pathway | ↑ Body weight and renal index ↓ Tubular epithelial cell apoptosis ↓ Expression of GRP78, CHOP and Caspase-12 |
– | Sprague Dawley Rats | [41] |
| Etoricoxib | Anti-inflammatory | ↓ Inflammation (iNOS) ↓ Apoptosis (BAX) No changes to creatinine, BUN, GSH, and MDA |
– | Rats | [40] |
| Eugenol | Antioxidant and anti-inflammatory properties | ↓ sCr and BUN ↓ PAS tubular injury score ↓ cytoplasmic vacuolization of proximal tubular cells |
– | BALB/c mice | [42] |
| Ferrostatin-1 | Inhibits Ferroptotic cell death | ↓ sCr and BUN ↓ apoptosis (TUNEL stain) ↓ Tubular injury score (H&E) ↓ Lipid peroxidation |
– | C57BL/6J mice | [43] |
| Isoorientin | Anti-inflammatory, antioxidant | ↓ ROS generation ↓ Apoptosis ↓ Inflammation |
mTECs | Nrf2−/− | [44] |
| Monotropein | Antioxidant, anti-inflammatory and antiapoptotic | ↓ Tubular injury ↓ markers of oxidative stress ↓ markers of apoptosis ↓ BUN, no reduction in sCr |
– | BALB/c mice | [45] |
| Paricalcitol | Synthetic vitamin D deficiency | ↓ MDA (HK-2 cells) ↓ Cell death (HK-2 cells) ↓ sCr and BUN (WT mouse) ↓ Tissue Injury (WT mouse) |
HK-2 cells | WT mice | [43] |
| Quercetin | Anti-inflammatory | ↓ sCr and BUN ↓ mRNA expression of IL-1β, IL-6, TNF-α ↓ reduced tubular necrosis score ↓ activity of Syk/NF-κB |
– | C57BL/6J mice | [46] |
Table 2. Genetic deletion studies investigating in vivo mechanisms associated with inflammatory processes involved in cisplatin-induced AKI pathogenesis. Key genetic deletion studies that have been targeted in both recent and hindsight applications targeting cisplatin-induced AKI with majority focusing on inflammatory pathways.
| Genetic Deletion | Mechanism of Action | Results | Knockout Model | Reference |
|---|---|---|---|---|
| CXCL16 | Antiapoptosis and anti-inflammatory | ↓ Apoptosis of tubular cells ↓ Caspase-3 activation ↓ inhibition of macrophage and T cell infiltration |
CXCL16 −/− mice C57BL/6J background WT | [47] |
| CYP2e1 | Antioxidant | ↓ ROS ↓ BUN ↓ sCr ↑ creatinine clearance |
CYP2e1 −/− mice 129/sv background WT | [48] |
| IL-6 | Antioxidant | ↑ 4-HNE ↓ SOD1 ↓ SOD2 (no significance) ↑ ERK phosphorylation ↑ COX-2 |
IL-6 −/− mice C57BL/6J background WT | [49] |
| IL-33 | Pro-inflammatory | ↑ BUN ↑ sCr ↑ NGAL No attenuation in ATN and tubular apoptosis scores ↓ tumor weight, volume, and growth ↑ Cisplatin efficacy |
IL-33 −/− mice C57BL/6J background WT | [50] |
| NLRP3 | Unknown | No change to BUN, sCr, ATN score and tubular apoptosis score. | NLRP3 −/− mice C57BL/6J background WT | [51] |
| PARP-1 | Anti-inflammatory, antioxidant and antinitrative | ↓ BUN ↓ sCr ↓ PAS tubular injury score |
PARP-1 −/− mice C57BL/6J background WT | [52] |
| T cell | Pro-inflammatory | ↑ cisplatin administration survival rate ↓ sCr ↓ tubular injury score ↓ TNF-α |
nu/nu mice | [53] |
| TAK1 | Antiapoptotic, Anti-inflammatory | ↓ Apoptosis of tubular cells ↓ Caspase-3 activation ↓ reduced mRNA expression of IL-6, TNF-α, MCP-1 and MIP-2 ↓ JNK phosphorylation |
PT-TAK1 −/− mice | [54] |
| TLR-2 | Inflammatory response | ↑ BUN ↑ sCr ↑ tissue injury score |
TLR2 −/− | [55] |
| TLR4 | Anti-inflammatory response | ↓ BUN ↓ sCr ↓ tissue injury index ↑ IL-4 and IL-10 |
TLR4 −/− | [55] |
| TLR-9 | Pro-inflammatory | No significant change to either serum urea or tubular injury score. | TLR-9 −/− | [56] |
| TNF-α | Potentially anti-inflammatory | ↓ BUN ↓ tubular necrosis score |
TNF-α −/− | [33] |
2. Pharmacological Approaches Targeting Cisplatin Cellular Uptake
2.1. Cellular Uptake Transporters of Cisplatin
The cellular uptake of cisplatin has been implicated in the pathogenesis of CIAKI. Organic cation transporter 2 (OCT2), copper transporter 1 (CTR1), and the less explored volume-regulated anion channels (VRAC) are involved in cisplatin transportation into kidney cells [38] by enabling platinum accumulation, which has been linked to kidney dysfunction [28] (Figure 2). Kidney tissue following cisplatin treatment showed a five-fold increase in cisplatin concentration compared to serum, indicative of PTEC accumulation [9]. Organic cation transporter 2 is one of the transporters affiliated with cisplatin cellular uptake.
2.2. Organic Cation Transporter 2 (OCT2)
OCT2 is expressed on the basolateral membrane of PTEC [38][40][41] and plays a central role in cisplatin uptake into tubular cells [38][40][41]. Amongst the transporters responsible for CIAKI, it has been shown that 30% of nephrotoxic effects caused by cisplatin is directly mediated by OCT2 uptake [49]. In in vitro studies investigating OCT2-mediated cisplatin cellular uptake, pharmacological inhibition was noted of OCT2 by cimetidine-inhibited cisplatin-induced apoptosis. In addition, nephrotoxicity stimulated by cisplatin transportation into renal tubular cells and subsequent platinum accumulation could be decreased with orally administered imatinib (a tyrosine kinase inhibitor) in rats. Histological investigations of kidney tissue confirmed that there was no evidence of severe renal damage in mice co-treated with cisplatin and imatinib. However, tubular degeneration was observed in cisplatin-treated groups [42]. The results of blood analysis (plasma urea, nitrogen, creatinine, and creatinine clearance) were indicative of improved kidney function and platinum accumulation following imatinib adjunct therapy. In OCT2-expressed HEK293 cell studies, adjunct administration of cisplatin and imatinib showed decreased accumulation of platinum in PTECs and decreased cisplatin-induced cytotoxicity [42]. However, despite the renoprotective effects observed in preclinical animal models, imatinib has not provided positive toxicology results. According to the US Food and Drugs Administration adverse reporting system, 44 imatinib-treated cases cited renal-related toxicity. Of these 44 cases, 25 manifested as AKI [50]. As such, this may not be an adequate clinical treatment. Potentially irreversible acute kidney injury was also observed in a nonclinical trial in imatinib-treated chronic myeloid leukemia patients [51].An experimental study using OCT2-deficient mice showed impairment of cisplatin uptake in renal cells, evident by reduced platinum accumulation [41]. Cairimboli et al. confirmed the importance of OCT2 in cisplatin uptake [39]. The authors associated the overexpression in HEK293 cells with increased cisplatin uptake causing cisplatin toxicity, because of increased cellular sensitivity [39]. To date, many pharmacological approaches targeting molecules responsible for cisplatin uptake or transportation into PTECs have been explored [38][42][52][53][54][55]. A murine model of CIAKI demonstrated downregulation of OCT2 expression by formononetin inhibited the development of AKI associated with cisplatin treatment through stimulation of renal tubular cell proliferation, survival, and apoptosis inhibition [52]. Despite the ameliorating effects of in vivo OCT2 inhibition in murine models of CIAKI, human studies failed to display the same renoprotective effects. Fox and colleagues used a randomized crossover experimental design to assess the prevention of cisplatin-induced nephrotoxicity using the OCT2 inhibitor pantoprazole, in young patients with osteosarcoma. To assess the effects, novel biomarkers were tested to investigate glomerular and tubular function. Measurement of serum cystatin c was used as an indirect indicator of GFR, and urinary biomarkers N-acetyl-β-glucosaminidase (NAG), kidney injury molecule-1 (KIM-1), and neutrophil gelatinase-associated lipocalin (NGAL) were used to quantify the degree of renal injury caused by cisplatin. The results of this study showed that concurrent administration of cisplatin with pantoprazole provided no protection against renal injury or function in young cancer patients [56]. Interestingly, a more recent study showed that pantoprazole can ameliorate CIAKI in mice [43]. Given the contradictory results of OCT2 inhibition on CIAKI in animal-versus-human studies further research needs to be conducted. It is important to note that OCT2 murine models were nontumor bearing, whilst the human studies were conducted in cancer patients, which could be a contributing factor to the failed clinical study.
2.3. Copper Transporter 1
CTR1 is located on the basolateral membrane of proximal tubules and is highly expressed in human kidneys [38][40]. The exact role of CTR1 in cellular uptake of cisplatin into renal proximal tubules resulting in nephrotoxicity is incompletely understood. However, studies have shown that CTR1 downregulation is protective against platinum accumulation [38]. The knockdown of CTR1 reduces cisplatin nephrotoxicity by up to 80% in both mouse embryonic fibroblasts and yeast [53][55]. In vivo studies indicated elevated levels of CTR1 expression was associated with increased cisplatin accumulation in tumors, a process also observed in PTEC [38][57]. Pabla and colleagues investigated the relationship between cisplatin and CTR1 expression to further define the role that CTR1 plays in nephrotoxicity. Interestingly, in mice, there were no significant differences in CTR1 expression 1–3 days following cisplatin treatment. They also demonstrated that incubation of HEK293 cells with copper generated both monomeric and trimeric CTR1 knockdown, resulting in approximately 50% diminution in cisplatin accumulation and a 30% reduction in apoptosis. Furthermore, CTR1 knockdown cells incubated with the OCT/MATE inhibitor cimetidine further inhibited both cellular uptake and apoptosis following treatment with cisplatin [38][39]. The results of this study have shown that although both CTR1 inhibition and OCT2 inhibition alone are options to prevent nephrotoxicity, the combination of CTR1 and OCT2 inhibition together has better therapeutic potential. Interestingly, the majority of cellular uptake research regarding cisplatin into renal cells has focused on the two major cisplatin transporters OCT2 and CTR1. However, there have also been suggestions that there are other entry points for cisplatin into renal cells that are yet to be explored in models of CIAKI such as VRAC channels. Reduced VRAC channel activity is associated with cisplatin resistance [58], and the presence of VRAC channels in kidney cells [59] highlights a potential avenue for CIAKI research. Additionally, impaired cisplatin efflux has been shown to contribute to cisplatin accumulation and the nephrotoxic effects that follow [46][60].
2.4. OAT1/OAT3
In addition to OCT2 and CTR1, the organic anion transporter (OAT) family(OAT1 and OAT3 have also shown to transport cisplatin and potentially a nephrotoxic metabolite into PTEC resulting in nephrotoxic injury to renal cells [44]. OAT transporters are largely concentrated in the basolateral membrane of PTEC and facilitate transportation of hydrophilic anions into cells. This intake is via secondary/active transportation responsible for regulating anion balance in the body [61]. To investigate the influence of OAT transporters on cisplatin-induced nephrotoxicity, C57BL/6J mice with genetic deletion of OAT1 and OAT3 were injected with 30 mg/kg cisplatin. In cisplatin-treated wildtype mice, there were increases in biomarkers of CIAKI, in addition to histological indication of kidney damage such as tubule dilation and necrosis. There was no evidence of kidney dysfunction in OAT1- and OAT3-deficient mice treated with cisplatin. Further studies are needed to further understand the role of each individually and the interaction they have together on CIAKI nephrotoxicity. A different model was used to investigate OAT-stimulated CIAKI using nilotinib. Nilotinib is a tyrosine kinase inhibitor shown to noncompetitively inhibit OCT2 and both OAT1 and OAT3 [44]. Nilotinib was given to OCT1/2 −/− mice simultaneously with cisplatin, with results indicating no loss of kidney function in the adjuvant cisplatin- and nilotinib-treated group as confirmed by reduced BUN levels. This indicates that OAT1/inhibition by nilotinib provides some evidence of amelioration of CIAKI; however, as the paper elucidates, further investigations in the mechanisms of mitigation are required [44]. A separate study investigated the effects of nilotinib in a rodent model of CIAKI. Male Wister albino rats were treated with 25 mg/kg Nilotinib 4 days prior to a single intraperitoneal injection of 6 mg/kg cisplatin and 6 days following the cisplatin injection. Results of their study showed that nilotinib improved creatinine clearance compared to cisplatin-treated rats; however, it had no influence on increased BUN [45]. It was also observed that nilotinib attenuated cisplatin increase in MDA, a biomarker of oxidative stress [62]. Morphological changes showed amelioration of CIAKI by nilotinib. Although this study did not look specifically at nilotinib influence on OAT1 and OAT3, it does confirm its ability to prevent CIAKI. Given this information, there is a clear correlation and link between the transporters, and therefore, further investigations need to be undertaken to understand their interactions and the influence that has on mediating cisplatin uptake. Given cisplatin uptake is the initial step mediating its nephrotoxic effects, potentially inhibiting all three synergistically may be an ideal strategy for CIAKI prevention.
3. Cisplatin-Induced Acute Kidney Injury: Role of the Immune System
Inflammatory pathways have been linked to major pathophysiological mechanisms resulting in CIAKI [63][64][65][66][67][68]. Models of AKI resulting from ischemia, sepsis, and nephrotoxicity all were presented with structural and functional changes to the vascular or tubular endothelium. These changes attract immune cells that infiltrate damaged kidney tissue. Extensive research has implicated a large array of cytokines and chemokines into the robust inflammatory response observed in models of CIAKI (Figure 5).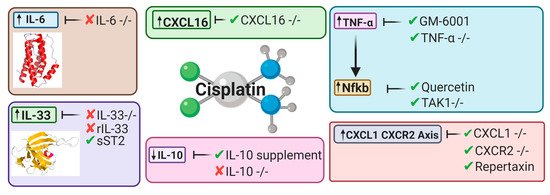 Figure 3. Key cytokines and chemokines upregulated or downregulated following cisplatin treatment. This diagram represents the therapeutic avenues published targeting inflammatory pathways and the targeted cytokines and chemokines involved [32][34][63][64][69][70][71][72][67].
Figure 3. Key cytokines and chemokines upregulated or downregulated following cisplatin treatment. This diagram represents the therapeutic avenues published targeting inflammatory pathways and the targeted cytokines and chemokines involved [32][34][63][64][69][70][71][72][67].
3.1. Toll-Like Receptors and Cisplatin-Induced Acute Kidney Injury
The innate immune system provides constituent primitive first-line defense mechanisms against an extensive repertoire of invading pathogens [73][74]. Integral to the establishment of innate immunity is a distinct class of pattern recognition receptors, referred to as toll-like receptors (TLRs) [75][76]. TLRs are evolutionary-preserved transmembrane type I sentinel glycoproteins, responsible for facilitating host surveillance through the identification of molecular signatures present on pathogens and self/host cells [75][76]. TLRs contain three structural components: (i) an intracellular C-terminal toll/interluikin-1 receptor domain (TIR), (ii) a central helix spanning the plasma or organelle membrane, and (iii) an ectodomain that extends into the extracellular environment or the lumen of the intracellular organelle [77][78]. The cytoplasmic domain is responsible for mediating signal transduction upon association with activating ligands [77][78]. The leucine-rich ectodomain provides diversity and specificity between individual TLRs, as each TLR can respond to different pathogenic (Table 3) and endogenous activating ligands [78][79]. TLR expression is abundant in human non-immune and immune tissues, including cardiac; pulmonary; nervous; hepatic; gastrointestinal; lymphoid; reproductive; and renal [73]. Interestingly, the human genome contains codes for 11 TLRs [80]. However, only 10 functional TLRs are expressed [80]. It has been suggested that the absence of TLR11 may be responsible for human susceptibility to urinary tract infections, as in murine models TLR11 provides resistance against uropathogenic Escherichia coli [80]. Activating ligands of TLRs include pattern-associated molecular patterns (PAMP) [76] and danger-associated molecular patterns (DAMP) [81][82]. PAMPs are defined as highly conserved, invariant motifs present on pathogens, that promote microbial survivability [73][83]. DAMPs are host-derived endogenous cytoplasmic or nuclear immunogenic alarmins that are liberated by damaged, stressed, and necrotic cells in the presence or absence of pathogenic infection to restore homeostatic balance [81][82][84]. Independent of the origin of the activating ligand, the resulting end product of sterile inflammation, produced through the myeloid differentiation factor-88 (MyD88)-dependent pathway (TLR1, 2, 4–10) [85] or the TIR-containing adapter-inducing interferon-β (TRIF)-dependent pathway (TLR3 and 4) [86], is ubiquitous among TLRs [87]. Activation of TLRs results in the production of inflammatory modulators, including cytokines, chemokines, interferons, and adhesion molecules, promoting the inflammatory response [88].Table 3. Summary of location and primary pathogens recognized by toll-like receptors.
| Toll-Like Receptor | Location | Primary Pathogen (s) |
|---|---|---|
| 1 | Extracellular | Gram-positive bacterium Fungus Mycobacterium |
| 2 | Extracellular | Fungus Gram-positive bacterium Mycobacterium |
| 3 | Intracellular | Double-stranded virus |
| 4 | Extracellular | Gram-negative bacterium |
| 5 | Extracellular | Flagellum |
| 6 | Extracellular | Gram-positive bacterium Fungus |
| 7 | Intracellular | Single-stranded virus |
| 8 | Intracellular | Virus |
| 9 | Intracellular | Bacterium |
| 10 | Extracellular | Gram-positive bacterium |
TLR expression is abundant in non-diseased human renal tissue [73][89][90][91][92][93][94][95][96][97][98][99][100][101][102][103] (Figure 4). Thus, chronic unregulated and unresolving TLR activation may be responsible for immunopathological consequences, as augmented TLR-induced inflammation and increased expression have been reported in a plethora of non-\infectious and autoimmune diseases that target the renal system, including nephrotoxicity [104]; renal disease [105][106][107]; lupus nephritis [92]; and diabetic nephropathy [108]. Furthermore, TLRs have also been implicated as potential drivers of cisplatin-induced pathologies and toxicities, including AKI [109][110][111]; renal injury [112][113][114]; allodynia [110]; and ototoxicity [115]. As demonstrated by TLR-deficient animal models [109][110][111] and polymorphisms in humans [116][117][118][119], the absence or improper function of TLRs may influence susceptibility and severity of pathologies affecting the renal system. Therefore, immunopathological consequences and renoprotective abilities originating from TLR activation in CIAKI are described.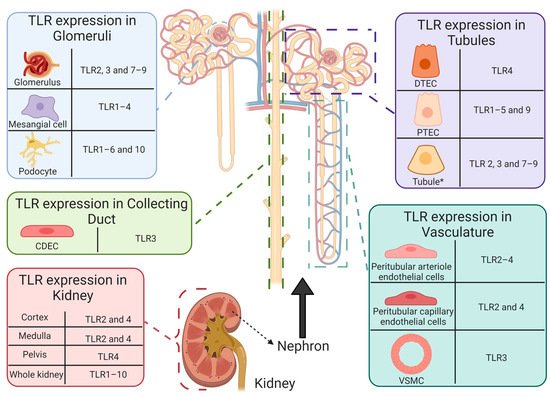 Figure 4. Functional expression of toll-like receptors in healthy human intrarenal cells and tissue. Basal expression of TLR1–10 has been reported in healthy human tissue [73]. However, determination of renal cell specific TLR expression remains limited. To date, TLR expression has been reported in intrarenal cells and structures, including: CDEC (TLR3 [89]), DTECs (TLR4 [90]), glomeruli (TLR2 [91]; TLR3 [89][92]; TLR7–9 [92][93]), mesangial cells (TLR1–4 [89][91]), peritubular arteriole (TLR2 [91] and TLR3 [89]) and capillary (TLR2 [91][94] and TLR4 [94]) endothelial cells, podocytes (TLR1–6 and 10 [95][96]), PTECs (TLR1–5 [94][97][98][99][100] and 9 [101]), tubules * (TLR2–4 [91][92][93][102] and TLR7–9 [93][103]), VSMCs (TLR3 [89]) and renal cortex (TLR2 [91] and TLR4 [102]), medulla (TLR2 [91] and TLR4 [102]), and pelvis (TLR4 [102]). * Tubules refers to literature that does not state the specific tubule on which TLR expression was reported. Abbreviations: collecting duct epithelial cell, CDEC; DTEC, distal tubule epithelial cell; PTEC, proximal tubule epithelial cell; toll-like receptor, TLR; vascular smooth muscle cell, VSMC.
Figure 4. Functional expression of toll-like receptors in healthy human intrarenal cells and tissue. Basal expression of TLR1–10 has been reported in healthy human tissue [73]. However, determination of renal cell specific TLR expression remains limited. To date, TLR expression has been reported in intrarenal cells and structures, including: CDEC (TLR3 [89]), DTECs (TLR4 [90]), glomeruli (TLR2 [91]; TLR3 [89][92]; TLR7–9 [92][93]), mesangial cells (TLR1–4 [89][91]), peritubular arteriole (TLR2 [91] and TLR3 [89]) and capillary (TLR2 [91][94] and TLR4 [94]) endothelial cells, podocytes (TLR1–6 and 10 [95][96]), PTECs (TLR1–5 [94][97][98][99][100] and 9 [101]), tubules * (TLR2–4 [91][92][93][102] and TLR7–9 [93][103]), VSMCs (TLR3 [89]) and renal cortex (TLR2 [91] and TLR4 [102]), medulla (TLR2 [91] and TLR4 [102]), and pelvis (TLR4 [102]). * Tubules refers to literature that does not state the specific tubule on which TLR expression was reported. Abbreviations: collecting duct epithelial cell, CDEC; DTEC, distal tubule epithelial cell; PTEC, proximal tubule epithelial cell; toll-like receptor, TLR; vascular smooth muscle cell, VSMC.
3.1.1. Toll-Like Receptor 2 is Protective in Cisplatin-Induced Acute Kidney Injury
TLR2 is unique, as it requires recruitment of other TLRs to form heterodimers (TLR1 [120]; TLR6 [120]; and TLR10 [121]) to facilitate activation and subsequent signal transduction, promoting inflammation. While it has been postulated that TLR2 can form a homodimer, it has yet to be observed [122]. The ability of TLR2 to form heterodimer complexes with other TLRs enables it to recognize a broad spectrum of pathogens (e.g., bacterium; fungi; helminth; mycobacterium; protozoa; and virus) [123] and host-derived endogenous ligands [124]. Previous studies involving CIAKI have reported a beneficial role of TLR2 [109][125] in the progression of disease development through mediated autophagy, which has been shown to protect renal cells from the detrimental effects of cisplatin-mediated cell death [126]. Exacerbated CIAKI has been reported in mice with TLR2 deficiency, receiving daily intraperitoneal injections of cisplatin (20 mg/kg/day) [125]. Within 24 h of receiving cisplatin, TLR2-deficient mice displayed significantly increased levels of sCr and BUN, and severe morphological changes to renal tissue, including loss of brush-border cells, tubule dilation, and cast formation [125]. Furthermore, TLR2-deficient mice had reduced renal tubular epithelial cell autophagy, associated with increased protein levels of p62 (an autophagy substrate chaperone, involved in delivering of ubiquitinated peptides to autophagosomes for proteasomal degradation [127]), and decreased levels of microtubule-associated protein 1A/1B light chain 3 II (LC3 II) (a LC3-phosphatidylethanolamine conjugate that is integrated into lipid membranes of autophagosomes and phagophores, which interacts with p62 to select targets for degradation [128][129]) and phosphorylation of phosphoinositide 3-kinase and protein kinase B [125]. This study also reported significantly increased mRNA and protein expression of TLR2 in wild-type mice 24 h after cisplatin treatment, which continued to increase in a time-dependent manner [125]. A complimentary study also reported a protective role of TLR2 during CIAKI, as the absence of TLR2, in mice treated with cisplatin (20 mg/kg), resulted in renal dysfunction shown by increased levels of sCr, BUN, and urea [109]. Histological assessment determined that mice with TLR2 deficiency developed exacerbated renal injury and structural damage, including enlargement of glomerular cavity, renal tubular dilation, renal epithelial cell detachment, formation of renal casts, and infiltration of inflammatory cells (macrophages and neutrophils) into the renal medulla and presence of necrosis [109]. Additionally, lack of TLR2 reduced survivability and failed to protect mice from cisplatin-induced weigh loss [109]. This study also demonstrated a protective role in renal autophagy during CIAKI through induction of the TLR2 pathway, as autophagosome forming molecules may depend on the TLR2 signal [109]. Renal tubular cells isolated from TLR2-deficient mice had decreased levels of autophagy genes (LC3 and autophagy-related 5) and proteins (LC3II, autophagy-related protein-5 and CCAAT-enhancer-binding protein-homologous protein) after cisplatin treatment [109]. Taken together, these results suggest that TLR2 plays a beneficial role during CIAKI by regulating autophagy and reducing renal dysfunction and pathological changes.
3.1.2. Toll-Like Receptor 4 Has a Detrimental Role in Cisplatin-Induced Acute Kidney Injury
Since its discovery in 1997 [130], TLR4 has been extensively investigated in the literature, and its structure, function, and signal transduction remain the most characterised and established of the TLRs [131]. TLR4 plays an essential role in host Gram-negative immunity by identifying lipopolysaccharides, a lipid and polysaccharide conjugate that is a major component in the outer membrane of Gram-negative bacteria [132]. However, the ability of TLR4 to recognize a repertoire of pathogens has been reported, including enveloped viruses [133] and viral proteins (viral fusion proteins [134] and glycoproteins [135]), Gram-positive bacteria [136], and helminths [137]. Furthermore, TLR4 can respond to a broad range of DAMPs, including endoplasmin [131], high-mobility group-1 [138], and heat-shock protein 70 [139], which have been shown to be upregulated during cisplatin treatment. Interestingly, TLR4 has also been shown to participate in transition metal sensing [140], and metals, including nickel, cobalt, and platinum [115][140] have been observed as TLR4-activating ligands. Due to the increase in circulating DAMPs and cisplatin (a platinum-based chemotherapy) acting as TLR4 ligands, TLR4 has been implemented as a major contributor in driving pathogenesis and development of CIAKI through upregulation of inflammation and proinflammatory and subsequent renal dysfunction, renal tissue injury, and nephrotoxicity [131]. A study involving TLR4-deficient mice administered a toxic dose of cisplatin (20 mg/kg) to induce acute renal failure within 72 h and reported significantly reduced markers of inflammation, nephrotoxicity, and renal function and decreased renal injury and histological abnormalities [131]. To determine the effect that TLR4 deficiency has on cisplatin-induced renal dysfunction and structural changes, BUN and sCr were used as indicators of function [131]. Severe renal failure (indicated by elevated levels of BUN and sCr) was observed between 48 and 72 h in WT after bolus dose of cisplatin and was accompanied by histological abnormalities including advanced tubular injury, cast formation, absence of brush-border membranes, shedding of tubular epithelial cells, necrosis of renal tubule cells, and dilation in renal tubules [131]. Mice with TLR4 deficiency has significantly preserved renal function 72 h after administration to cisplatin, as shown by reduced BUN ad sCr concentrations and minimal histological changes. Thus, indicating that TLR4 contributes to structural and functional consequences during CIAKI [131]. Furthermore, immunopathological consequences, including reduced leukocyte infiltration, decreased concentrations of cytokine and chemokine in serum (i.e., TNF-α; IL-1β; IL-2; IL-6; and IL-10), kidney (i.e., TNF-α; IL-6; CCL5; MCP-10; and KC) urine (i.e., TNF-α; IL-2; IL-6; CCL5; MCP-1; KC; and IP-10), and reduced activity of p38 MAPK and JNK phosphorylation was also observed in TLR4-deficient mice when compared to WT [131]. Thus, suggesting that the potent inflammatory response initiated by TLR4 may be responsible for initiating CIAKI [131]. Therefore, a tailored therapy encompassing a combination of cisplatin and a TLR4 inhibitor is an appealing approach to preserve renal structural integrity and preservation of function in CIAKI.A potential TLR4 inhibitor to be used in conjunction with cisplatin, which has shown promising results in septic-induced AKI, is resatorvid (TAK242) [141][142]. TAK242 is a cyclohexene derivative [143], which exerts its inhibitory effect by binding to the intracellular domain of TLR4. Upon binding, TAK242 causes a confirmation change in the cytoplasmic tail, which results in the inability of the bridging adaptor molecules TIR-containing adapter protein/myeloid differentiation factor-88 and translocating chain-associated membrane protein/TRIF to associate [144], thus preventing TLR4 signal transduction and subsequent production and release of proinflammatory mediators [144]. Administration of TAK242 to ovine models of Gram-negative bacteria resulted in enhanced renal function, demonstrated by abolishment of impaired Cr clearance in the urine, reduced BUN and sCr, prevention of renal hypoperfusion, and reduced swelling of endothelial cells in glomerular capillaries [141][142]. Additionally, a recent article has shown that TAK242 is able to enhance the cytotoxic effect of cisplatin in breast and ovarian cancer cells, while preventing its toxic effects of cells [145]. Thus, suggesting that dual treatment with TAK242 and cisplatin could enable a reduced dose of cisplatin given to patients. Taken together, TAK242 should be further investigated in CIAKI, as it represents a pharmaceutical that could (i) prevent detrimental chronic inflammation, (ii) retain structural integrity and renal function, and (c) intensify the effect of cisplatin in CIAKI.
3.1.3. Toll-Like Receptor 9 is Protective in Cisplatin-Induced Acute Kidney Injury
TLR9 is a cytosolic receptor bound to the membranes of intracellular organelles (e.g., endosomes, lysosomes, and endolysosomes) and is responsible for identification of unmethylated cytosine-phosphate-guanosine DNA present in bacteria and viruses [101][146][147]. Furthermore, TLR9 has been shown to directly contribute to cardiac, hepatic, and renal ischemic tissue injury [101] and lupus nephritis [148][149] through recognition of endogenous mitochondrial DNA products [150], chromatin IgG complex [151], GP96 [152], high-mobility group box-1 [153], and heat-shock protein 90 [154]. A study involving CIAKI determined a protective immunomodulating role of TLR9 in disease progression, as TLR9 absence resulted in accelerated and increased pathological development [105]. TLR9-deficient mice were administered cisplatin (20–25 mg/kg) via intraperitoneal injection and were then euthanised 24–72 h after treatment [105]. When compared to wild-type controls, TLR9-deficient mice had greater renal dysfunction and histological injury 24 h after cisplatin exposure, as shown by significant increases in serum urea, tubular injury score, neutrophil, and CD4+ cells and mRNA expression of CXCL1/2 [105]. Additionally, augmented disruption of renal tubular structure and integrity, tubular necrosis, cast formation, and accumulation of tubular cell debris was observed in kidneys derived from TLR9-deficient mice when compared to wild-type controls [105]. However, after 56 h of cisplatin exposure, no significance in renal function and histological injury was observed between TLR9-deficient mice and wild-type controls, suggesting that increased renal injury was not mediated by effector cell function in TLR9-deficient mice [105]. Previous in vitro studies involving human cells have postulated simultaneous activation of regulatory T cells (Tregs) and TLR9, suggesting the ability of TLR9 activation to regulate/suppress Treg activity [155]. Therefore, using this hypothesis, the authors determined if exacerbated renal injury and dysfunction was caused by Treg cell activity modulated by TLR9 [105]. The findings from this study showed that TLR9-deficient Tregs were not defective in functionally, abundance or apoptotic-inducing abilities but that the amount of adhesion molecules responsible for Treg recruitment into the kidneys was reduced in TLR9 absence [105]. This is supported by literature that shows that Tregs must be actively recruited into renal tissue to exert renoprotective abilities in AKI [105][156]. Therefore, taken together, this study suggests that TLR9 plays a beneficial role in CIAKI by enabling recruitment of Tregs into inflamed renal tissue [105].
3.2. Cytokines
3.2.1. Tumor Necrosis Factor Alpha
Cisplatin-induced nephrotoxicity involves the activation of a proinflammatory response [157]. Cisplatin has been linked to the increased expression of TNF-α in both serum and urine concentrations of AKI [31]. TNF-α is a branch of the TNF family. These proteins are important cytokines responsible for cell signaling. They play an important role in immunity as well as the possession of proinflammatory properties [15][158]. TNF-α inhibitors have shown that in the absence of TNF-α.α cisplatin-induced nephrotoxicity is attenuated, indicating that TNF-α plays a significant role in cisplatin’s nephrotoxic effects. Treatment with the matrix metalloprotease inhibitor GM-6001, a TNF-α antagonist, reduced urea levels in GM-6001 mice compared to cisplatin, showing treatment with GM-6001 enhanced renal function. This improvement in renal function correlates with the improved renal histology [33]. Genetic modification of TNF-α also displayed renoprotective effects. Silencing of TNF-alpha showed amelioration of kidney dysfunction resulting from cisplatin treatment, with TNF-alpha −/− mice protected against the nephrotoxic effects of cisplatin [33]. This highlights a crucial role of proinflammatory cytokines and chemokines in the pathogenesis of CIAKI, particularly a central role for TNF-alpha [33]. Interestingly, TNF-α enhances the anticancer properties of cisplatin in breast cancer cells, both in vitro and in vivo [159]. It would be interesting to investigate the effects of TNF-α downregulation on the cytotoxicity of cisplatin in an in vivo model of CI-AK. In addition to upregulation by cisplatin, Nfκb is also activated by TNF-α [160] and in CIAKI [33].
3.2.2. Nuclear Factor Kappa-Light-Chain Enhancer of Activated B Cells
Nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) is a key prosurvival and inflammatory cell transcription factor and is induced by cisplatin treatment [161]. Expression of Nfκb was significantly upregulated in rats treated with cisplatin compared to control rats. SA pretreatment significantly downregulated NF-kB expression compared to cisplatin [162]. Interleukin-6 (IL-6) and TNF-α levels were markedly reduced in the SA and cisplatin compared to only cisplatin-treated rats; however, levels were not completely restored to control levels. Quercetin is a flavonoid that elicits anticancer, anti-inflammatory, and antioxidant properties. Results of this study showed quercetin restored kidney function indicated by reduced serum BUN and creatinine compared to cisplatin-treated mice. Quercetin also downregulated mRNA expression of key inflammatory markers IL-1β, IL-6, and TNF-α compared to control, indicated by real-time PCR. Western blot analysis indicated Quercetin treatment decreased Syk/NF-κB activity in kidney tissue following cisplatin-induced AKI. They concluded that inhibition of Mincle/Syk/NF-κB signaling by quercetin exerts its renoprotective effects through reduced inflammation [64]. The progression would be to test quercetin in a tumor bearing model of cisplatin-induced AKI, and it is one of the first anticancer drugs to be used and could potentially exert its renoprotective effects whilst maintaining cisplatin’s cytotoxicity. This is a promising therapeutic option for cancer patients. In addition to pharmacological inhibition of NF-kB displaying renoprotective effects, transcriptional inhibition of NF-kB also protects against cisplatin-induced AKI [161]. The literature has investigated the role of transforming growth factor-β-activated kinase-1 (TAK1) in cisplatin-induced acute kidney injury [163][164]. TAK-1 is a component of the NF-kB pathway and protects cells from TNF-α-induced cell death through upregulation of antiapoptotic proteins [165]. TAK1 expression is known to be upregulated in response to cisplatin treatment [164]. TAK1 gene disruption was performed in order to generate a knockout in the proximal tubule to investigate the role of TAK1 following cisplatin treatment. Lower sCr and BUN and levels were observed in TAK1-deficient mice 72 h following injections of 20 mg/kg of cisplatin compared to controls. This is indicative of reduced renal dysfunction in the TAK-1-deficient group. H&E staining of kidney sections investigating tubular epithelial cell injury, proximal tubular dilation, and cast formation showed TAK-1 deficiency reduced histological evidence of kidney injury [163]. Following this knockout study, a pharmacological study of cisplatin-induced AKI was performed using a TAK1 inhibitor. Results of the study determined that TAK-1 inhibition increased sCr and BUN, suggesting TAK-1 plays a protective role against CIAKI. Interestingly, following the increased expression of TNF-alpha and consequently Nfκb observed in CIAKI, this is often correlated with a downregulation in Interleukin-10 (IL-10).
3.2.3. Interleukin-10
There is significant evidence implicating proinflammatory cytokines in the pathogenesis of cisplatin-induced renal dysfunction [166][167][168], stimulating the focus on anti-inflammatory cytokines as therapeutic targets against CIAKI. IL-10 is an anti-inflammatory cytokine produced by T helper cells, T cells, dendritic cells, and macrophages [31]. IL-10 inhibits many physiological processes including early-phase inflammation, cytokines, chemokines, neutrophil activation, and NO production [167]. Deng and colleagues conducted a study to investigate the effects of IL-10 on CIAKI in male BALB/c and C57BL/6 mice. A histological analysis was conducted 72 h after mice were treated with cisplatin. Results showed evidence of necrosis in the renal proximal tubule and the straight tubule. Cast formation and leukocyte accumulation was also shown, indicative of an inflammatory response. The results of the study showed that serum concentrations of IL-10 were downregulated in response to cisplatin treatment and maximal inhibition of renal injury was observed when cisplatin and 1g of IL-10 (determined via dose-dependent study) was administered concurrently. It was also determined that IL-10 administration one hour post cisplatin treatment also decreased renal damage [167]. A separate study in IL-10 KO mice showed enhanced CIAKI, further explicating its renoprotective effects [169]. Increased IL-10 expression has also been associated with the renoprotective effects of AT2 receptor stimulation following TLR4-induced inflammation [170]. This shows that enhanced expression of IL-10 is renoprotective and finding a pharmaceutical activator of IL-10 to assess in a model of AKI may provide therapeutic benefit. Tumor cells are known to overexpress key cytokines, such as IL-10. IL-10 protected against chemotherapeutic agent effects, through upregulation of antiapoptotic proteins such as Bcl-2 and Bcl-xL [171]. As such, further investigations into the effects of IL-10 attenuation of CIAKI in a tumor-bearing model should be completed. IL-33 another cytokine has been suggested to contribute to cisplatin’s nephrotoxicity.
3.2.4. Interleukin-33
IL-33 is a proinflammatory cytokine. This cytokine contains a receptor ST2, which attracts immune cells such as CD4+ and T cells via chemotaxis [31]. Elevated kidney expression of IL-33 was observed in CIAKI and tubular injury, suggesting that IL-33 may contribute to the nephrotoxic effects detected following cisplatin treatment [172][173]. Akcay and colleagues conducted a study using a decoy receptor to identify the role IL-33 and CD4+ T cells play in cisplatin-induced acute tubular necrosis (ATN) and apoptosis. In the study, two experimental groups were used. The first group was administered sST2, a decoy receptor to inhibit IL-33 function. It was found that when this was administered, a reduction in the infiltration of CD4+ T cells was observed along with decreased ATN and apoptosis. In the second group, recombinant IL-33 was administered and was found to intensify CIAKI, whilst in CD4+-deficient mice administered with IL-33, kidney structure and function remained unaffected. The results of this inferred that CD4+ T cells were accountable for the damage caused by IL-33; in addition they concluded that IL-33 inhibition may have a therapeutic potential in CIAKI [173]. Ravichandran and colleagues conducted a study that investigated the protective role of IL-33 deficiency on cisplatin-induced AKI. The results showed that IL-33 deficiency was not protective against CIAKI in mice [172]. The interesting component of this study that differs from most CIAKI models is that it was a tumor-bearing model. This stimulated investigations regarding not only the nephroprotective effects of IL-33 deficiency but also the effects of the deficiency on cisplatin’s anticancer activity. BUN and sCr were elevated in both IL-33 deficiency and WT mice treated with cisplatin in addition to increased expression of the AKI biomarker NGAL. ATN and tubular apoptosis observed in cisplatin-induced AKI were not alleviated in IL-33-deficient mice, furthering the evidence that IL-33 deficiency could not attenuate cisplatin’s nephrotoxic effects. It has been demonstrated that IL-33 is upregulated in cisplatin treatment [173]. Although IL-33 may be involved and contribute to CIAKI, it is unlikely to be causative. To investigate IL-33 deficiency on the cytotoxicity of cisplatin, cleaved, caspase-3 expression was examined. In tumors of vehicle, WT-treated and IL-33-deficient mice, no significant differences in caspase-3 expression was observed; however, caspase-3 levels were elevated in tumors of cisplatin WT mice but not in IL-33-deficient mice. Although there was evidence of reduced apoptosis, tumor weight, tumor volume, and tumor growth were all reduced in IL-33-deficient mice, a beneficial outcome for cancer prognosis [172]. Recent publications have implicated tumors themselves in the pathogenesis of AKI potentially through tumor lysis syndrome (TLS). TLS is caused by cancer treatments; however, they can occur spontaneously themselves, discharging cancer contents into the bloodstream, clinically presenting as hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia which can manifest and result in renal dysfunction [174]. Most publications to date use noncancer-bearing models to assess interventions for CIAKI. Healthy mice are injected with cisplatin either prior, concurrently, or following the intervention. This therefore investigates nephrotoxicity caused by cisplatin itself; however, it excludes the influence of cancer on kidney dysfunction. It has been suggested that TLS from cancer patients may contribute to the pathogenesis of CIAKI; therefore, Ravichandran and colleagues repeated the same experiments on IL-33 deficiency in cisplatin-treated mice without cancer. The results of this concluded that cancer did not influence the lack of renoprotection, elicited by IL-33 deficiency [172]. Knowing this, it highlights the importance of trialing therapies in both tumor-bearing and tumor-absent models of CIAKI.
3.2.5. Interleukin-6
IL-6 is a pleiotropic cytokine predominantly exerting proinflammatory functions but also exhibits anti-inflammatory properties [175]. It is a member of the interleukin family of cytokines, a collective whose involvement has been extensively researched in the pathology of CIAKI for many years. Serum and urine levels of IL-6 are elevated following cisplatin treatment, and it has been used as an early serum and urine biomarker of AKI [176]. However, IL-6 stimulation has been proposed to play a protective role in models of cisplatin-induced AKI implied to be via upregulation of antioxidant markers [69]. IL-6 −/− mice were administered 30 mg/kg of cisplatin via intraperitoneal injection. Twenty-four and 72 h post cisplatin administration blood and kidneys were harvested and analyzed for oxidative and antioxidative stress markers. Western blotting of IL-6 −/− cisplatin-treated mice showed increased expression of 4-HNE compared to wild-type mice. Following this, gene expression of free radical scavengers SOD1 and SOD2 in cisplatin-treated kidneys was analyzed via RT-PCR assays. Kidneys excised 24 h after cisplatin treatment yielded no significant difference in SOD1 expression; however, SOD1 expression was reduced in wild-type and IL-6 −/− mice 72 h after cisplatin treatment. In addition to investigating gene expression, enzymatic activity of SOD was analyzed indicating that SOD activity was significantly reduced in kidneys of IL-6 −/− mice compared to wild type [69]. Previously, it was determined that although IL-6 deficiency did not accelerate the development of systemic injury, it did accelerate progression of cisplatin-induced acute renal failure [177]. This indicates that IL-6 may play a protective role in CIAKI, but to date, there is no pharmacological IL-6 antagonism studies investigating this. Cancer models have showed that increased expression of IL-6 enhances acquired cisplatin resistance and reduces cytotoxicity; therefore, direct stimulation of IL-6 over-expression may reduce the efficacy of cisplatin [178] and therefore may not be an appropriate therapeutic target for cancer patients to prevent CIAKI. Many cytokines have been explored in models of CIAKI; however, recent literature has elucidated the role of specific chemokines in the pathogenesis of the disease.
3.3. Chemokines
The development and progression of an inflammatory response has long been associated with kidney damage and nephrotoxicity stimulated by cisplatin treatment [179]. Evidence has presented a role for proinflammatory chemokines as potential mechanisms associated with CIAKI, which is suggested to be mediated via TNF-α [33]. Both CXC and CC chemokines have varying roles in cancer, with many members from both subfamilies recruited to tumor sites playing either a pro- or anti-tumor role. Multiple chemokines from the CXC family have been linked to the pathogenesis of cancer, particularly in the stimulation of angiogenesis [180]. Several chemokine family members have also been shown to contribute to the pathogenesis of CIAKI.
3.3.1. CXCL16
CXC chemokine ligand 16 (CXCL16) is a member of the CXC family of chemokines [181]. CXCL16 has been associated with proinflammatory properties in multiple diseases [182][183]. Research suggests that CXCL16 may play a role in acute coronary syndrome most often associated with atherosclerosis [184]. Inhibition of CXCL16 has been linked to ameliorating effects in a variety of inflammatory related diseases such as liver inflammation and steatohepatitis [185], anti-GBM glomerulonephritis [186] and the regulation of CIAKI [181]. CXCL16 expression is upregulated in kidneys following cisplatin treatment [181]. To further understand the mechanisms stimulating this upregulation and the role CXCL16 plays in inflammation and apoptosis in renal tubular cells, wild-type and knockout mice were injected with 20 mg/kg of cisplatin. Seventy-two h after cisplatin treatment, the mice were culled. The results showed that kidney dysfunction was observed in wild-type mice, indicated by elevated levels of serum BUN. CXCL16 knockout mice were protected from cisplatin-induced renal dysfunction with reduced levels of serum BUN. Knockout mice also presented with reduced histological damage of renal tissue compared to WT mice. This study used RT-PCR to investigate mRNA expression of key proinflammatory cytokines associated with CIAKI. Results showed that following cisplatin injection expression of TNF-a, IL-1β, IL-6, and transforming growth factor β1 were upregulated in wild-type mice compared to the vehicle. Inhibition of mRNA expression of these molecules was observed in CXCL16 knockout mice. Decreased caspase-3 activation was also seen in CXCL16 −/− mice illustrating CXCL16’s role in tubular epithelial cell apoptosis. [181]. A separate study also involving CXCL16 −/− mice investigated renal injury associated with salt-sensitive hypertension. The study showed that DOCA-salt-treated −/− mice showed reduced renal dysfunction, proteinuria, BUN, and fibrosis compared to wild-type mice [187]. The collection of these studies has highlighted a role for CXCL16 in the inflammatory response associated with renal injury. Given the information and results observed in these studies, the progression would be to assess concurrent treatment with cisplatin and the pharmacological inhibition of CXCL16 in the prevention of nephrotoxicity. Pharmacological treatment using a monoclonal, rat anti-mouse CXCL16 neutralising antibody has shown to reduce liver inflammation in chronic hepatic injury [188]. More relative to AKI, a study involving the pharmacological inhibition of CXCL16 using an antiserum generated against CXCL16 showed reduced progression of anti-GBM glomerulonephritis suggested to be through its role in leukocyte influx [186]. Despite the evidence linking CXCL16 to CIAKI, it is not the only CXC chemokine linked to the disease. The CXCL1-CXCR2 axis has also been shown to be involved in renal damage following cisplatin treatment.
3.3.2. CXCL1-CXCR2 Axis
The chemokine (C-X-C motif) ligand 1 (CXCL1) belongs to the CXC family of chemokines. The molecular structure of CXCL1 can be either a monomer or a dimer and is a highly potent CXCR2 receptor agonist [189]. The CXCL1-CXCR2 axis has been implicated in a variety of inflammatory diseases, specifically through its role in neutrophil recruitment and microbial death at sites of tissue injury [189][190]. A key component in the immune response triggered by AKI promoting renal injury is the accumulation of key leukocytes, such as neutrophils and monocyte/macrophages [191]. CXCL1 and its upregulation has been secondarily investigated in models of CIAKI specifically in IL-33-deficient mice [172]. A recent study published demonstrated the inhibition of the CXCL1-CXCR2 axis provided ameliorating effects against renal damage induced by CIAKI [35]. Following cisplatin treatment, a significant increase in kidney mRNA and protein expression for both CXCL1 and CXCR2 compared to control was observed, indicating that the CXCL1-CXCR2 axis plays a role in the nephrotoxic effects induced by cisplatin. Reduced sCr and BUN were observed in both CXCL1- and CXCR2-deficient mice, indicating axis silencing improves renal function and AKI induced by cisplatin. There was also evidence of reduced renal neutrophil infiltration, indicating the potential of a reduced immune response in both CXCL1- and CXCR2-deficient mice. Following the genetic deletion studies, pharmacological inhibition of CXCL1 and CXCR2 with repertaxin displayed ameliorating effects against CIAKI. The results of their study concluded that inhibition of the CXCL1-CXCR2 axis was renoprotective against CIAKI through inhibition of p38 and Nfκb. [35]. Following this, the use of selective pharmacological antagonists of CXCL1 to and CXCR2 is needed to observe their isolated role in the disease pathogenesis of CIAKI.
This entry is adapted from the peer-reviewed paper 10.3390/cancers13071572
References
- Potočnjak, I.; Marinić, J.; Batičić, L.; Šimić, L.; Broznić, D.; Domitrović, R. Aucubin administered by either oral or parenteral route protects against cisplatin-induced acute kidney injury in mice. Food Chem. Toxicol. 2020, 142, 111472.
- Huang, S.-J.; Huang, J.; Yan, Y.-B.; Qiu, J.; Tan, R.-Q.; Liu, Y.; Tian, Q.; Guan, L.; Niu, S.-S.; Zhang, Y. The renoprotective effect of curcumin against cisplatin-induced acute kidney injury in mice: Involvement of miR-181a/PTEN axis. Ren. Fail. 2020, 42, 350–357.
- Abd El-Kader, M.; Taha, R.I. Comparative nephroprotective effects of curcumin and etoricoxib against cisplatin-induced acute kidney injury in rats. Acta Histochem. 2020, 122, 151534.
- Chai, Y.; Zhu, K.; Li, C.; Wang, X.; Shen, J.; Yong, F.; Jia, H. Dexmedetomidine alleviates cisplatin-induced acute kidney injury by attenuating endoplasmic reticulum stress-induced apoptosis via the α2AR/PI3K/AKT pathway. Mol. Med. Rep. 2020, 21, 1597–1605.
- Kadir, A.; Sher, S.; Siddiqui, R.A.; Mirza, T. Nephroprotective role of eugenol against cisplatin-induced acute kidney injury in mice. Pak. J. Pharm. Sci. 2020, 33, 1281–1287.
- Hu, Z.; Zhang, H.; Yi, B.; Yang, S.; Liu, J.; Hu, J.; Wang, J.; Cao, K.; Zhang, W. VDR activation attenuate cisplatin induced AKI by inhibiting ferroptosis. Cell Death Dis. 2020, 11, 1–11.
- Fan, X.; Wei, W.; Huang, J.; Liu, X.; Ci, X. Isoorientin attenuates cisplatin-induced nephrotoxicity through the inhibition of oxidative stress and apoptosis via activating the SIRT1/SIRT6/Nrf-2 pathway. Front. Pharmacol. 2020, 11, 264.
- Zhang, Y.; Chen, Y.; Li, B.; Ding, P.; Jin, D.; Hou, S.; Cai, X.; Sheng, X. The effect of monotropein on alleviating cisplatin-induced acute kidney injury by inhibiting oxidative damage, inflammation and apoptosis. Biomed. Pharmacother. 2020, 129, 110408.
- Tan, R.Z.; Wang, C.; Deng, C.; Zhong, X.; Yan, Y.; Luo, Y.; Lan, H.Y.; He, T.; Wang, L. Quercetin protects against cisplatin-induced acute kidney injury by inhibiting Mincle/Syk/NF-κB signaling maintained macrophage inflammation. Phytother. Res. 2020, 34, 139–152.
- Liang, H.; Zhang, Z.; He, L.; Wang, Y. CXCL16 regulates cisplatin-induced acute kidney injury. Oncotarget 2016, 7, 31652.
- Liu, H.; Baliga, R. Cytochrome P450 2E1 null mice provide novel protection against cisplatin-induced nephrotoxicity and apoptosis. Kidney Int. 2003, 63, 1687–1696.
- Mitazaki, S.; Honma, S.; Suto, M.; Kato, N.; Hiraiwa, K.; Yoshida, M.; Abe, S. Interleukin-6 plays a protective role in development of cisplatin-induced acute renal failure through upregulation of anti-oxidative stress factors. Life Sci. 2011, 88, 1142–1148.
- Ravichandran, K.; Holditch, S.; Brown, C.N.; Wang, Q.; Ozkok, A.; Weiser-Evans, M.C.; Nemenoff, R.; Miyazaki, M.; Thiessen-Philbrook, H.; Parikh, C.R. IL-33 deficiency slows cancer growth but does not protect against cisplatin-induced AKI in mice with cancer. Am. J. Physiol. Ren. Physiol. 2018, 314, F356–F366.
- Kim, H.-J.; Lee, D.W.; Ravichandran, K.; Keys, D.O.; Akcay, A.; Nguyen, Q.; He, Z.; Jani, A.; Ljubanovic, D.; Edelstein, C.L. NLRP3 inflammasome knockout mice are protected against ischemic but not cisplatin-induced acute kidney injury. J. Pharmacol. Exp. Ther. 2013, 346, 465–472.
- Mukhopadhyay, P.; Horváth, B.; Kechrid, M.; Tanchian, G.; Rajesh, M.; Naura, A.S.; Boulares, A.H.; Pacher, P. Poly (ADP-ribose) polymerase-1 is a key mediator of cisplatin-induced kidney inflammation and injury. Free Radic. Biol. Med. 2011, 51, 1774–1788.
- Liu, M.; Chien, C.-C.; Burne-Taney, M.; Molls, R.R.; Racusen, L.C.; Colvin, R.B.; Rabb, H. A pathophysiologic role for T lymphocytes in murine acute cisplatin nephrotoxicity. J. Am. Soc. Nephrol. 2006, 17, 765–774.
- Zhou, J.; An, C.; Jin, X.; Hu, Z.; Safirstein, R.L.; Wang, Y. TAK1 deficiency attenuates cisplatin-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 2020, 318, F209–F215.
- Andrade-Silva, M.; Cenedeze, M.A.; Perandini, L.A.; Felizardo, R.J.F.; Watanabe, I.K.M.; Agudelo, J.S.H.; Castoldi, A.; Gonçalves, G.M.; Origassa, C.S.T.; Semedo, P. TLR2 and TLR4 play opposite role in autophagy associated with cisplatin-induced acute kidney injury. Clin. Sci. 2018, 132, 1725–1739.
- Alikhan, M.A.; Summers, S.A.; Gan, P.Y.; Chan, A.J.; Khouri, M.B.; Ooi, J.D.; Ghali, J.R.; Odobasic, D.; Hickey, M.J.; Kitching, A.R. Endogenous toll-like receptor 9 regulates AKI by promoting regulatory T cell recruitment. J. Am. Soc. Nephrol. 2016, 27, 706–714.
- Ramesh, G.; Reeves, W.B. TNF-α mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J. Clin. Investig. 2002, 110, 835–842.
- Qin, Y.; Stokman, G.; Yan, K.; Ramaiahgari, S.; Verbeek, F.; De Graauw, M.; Van de Water, B.; Price, L.S. cAMP signalling protects proximal tubular epithelial cells from cisplatin-induced apoptosis via activation of Epac. Br. J. Pharmacol. 2012, 165, 1137–1150.
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute kidney injury. Lancet 2012, 380, 756–766.
- Kellum, J.A.; Unruh, M.L.; Murugan, R. Acute kidney injury. BMJ Clin. Evid. 2011, 394, 1949–1964.
- Lameire, N.; Van Biesen, W.; Vanholder, R. Acute kidney injury. Lancet 2008, 372, 1863–1865.
- Hanif, M.O.; Bali, A.; Ramphul, K. Acute renal tubular necrosis. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2020.
- Rahman, M.; Shad, F.; Smith, M.C. Acute kidney injury: A guide to diagnosis and management. Am. Fam. Physician 2012, 86, 631–639.
- Dalal, R.; Bruss, Z.S.; Sehdev, J.S. Physiology, renal blood flow and filtration. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2019.
- Oh, G.-S.; Kim, H.-J.; Shen, A.; Lee, S.B.; Khadka, D.; Pandit, A.; So, H.-S. Cisplatin-induced kidney dysfunction and perspectives on improving treatment strategies. Electrolytes Blood Press. 2014, 12, 55–65.
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin Nephrotoxicity. Toxins (Basel) 2010, 2, 2490–2518.
- Bennis, Y.; Savry, A.; Rocca, M.; Gauthier-Villano, L.; Pisano, P.; Pourroy, B. Cisplatin dose adjustment in patients with renal impairment, which recommendations should we follow? Int. J. Clin. Pharm. 2014, 36, 420–429.
- Ozkok, A.; Edelstein, C.L. Pathophysiology of cisplatin-induced acute kidney injury. Biomed. Res. Int. 2014, 2014, 967826.
- Rosner, M.H.; Perazella, M.A. Acute kidney injury in the patient with cancer. Kidney Res. Clin. Pract. 2019, 38, 295.
- Ramesh, G.; Reeves, W.B. TNF-α mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J. Clin. Investig. 2002, 110, 835–842.
- Zhang, B.; Ramesh, G.; Norbury, C.; Reeves, W. Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-α produced by renal parenchymal cells. Kidney Int. 2007, 72, 37–44.
- Liu, P.; Li, X.; Lv, W.; Xu, Z. Inhibition of CXCL1-CXCR2 axis ameliorates cisplatin-induced acute kidney injury by mediating inflammatory response. Biomed. Pharmacother. 2020, 122, 109693.
- Soni, H.; Kaminski, D.; Gangaraju, R.; Adebiyi, A. Cisplatin-induced oxidative stress stimulates renal Fas ligand shedding. Ren. Fail. 2018, 40, 314–322.
- Ni, J.; Hou, X.; Wang, X.; Shi, Y.; Xu, L.; Zheng, X.; Liu, N.; Qiu, A.; Zhuang, S. 3-deazaneplanocin A protects against cisplatin-induced renal tubular cell apoptosis and acute kidney injury by restoration of E-cadherin expression. Cell Death Dis. 2019, 10, 355.
- Pabla, N.; Murphy, R.F.; Liu, K.; Dong, Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am. J. Physiol Ren. Physiol. 2009, 296, F505–F511.
- Ciarimboli, G.; Ludwig, T.; Lang, D.; Pavenstädt, H.; Koepsell, H.; Piechota, H.-J.; Haier, J.; Jaehde, U.; Zisowsky, J.; Schlatter, E. Cisplatin nephrotoxicity is critically mediated via the human organic cation transporter 2. Am. J. Pathol. 2005, 167, 1477–1484.
- Ciarimboli, G. Membrane transporters as mediators of cisplatin side-effects. Anticancer Res. 2014, 34, 547–550.
- Ciarimboli, G.; Deuster, D.; Knief, A.; Sperling, M.; Holtkamp, M.; Edemir, B.; Pavenstadt, H.; Lanvers-Kaminsky, C.; Am Zehnhoff-Dinnesen, A.; Schinkel, A.H.; et al. Organic cation transporter 2 mediates cisplatin-induced oto- and nephrotoxicity and is a target for protective interventions. Am. J. Pathol. 2010, 176, 1169–1180.
- Tanihara, Y.; Masuda, S.; Katsura, T.; Inui, K.-I. Protective effect of concomitant administration of imatinib on cisplatin-induced nephrotoxicity focusing on renal organic cation transporter OCT2. Biochem. Pharmacol. 2009, 78, 1263–1271.
- Ismail, R.S.; El-Awady, M.S.; Hassan, M.H. Pantoprazole abrogated cisplatin-induced nephrotoxicity in mice via suppression of inflammation, apoptosis, and oxidative stress. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 1–11.
- Hu, S.; Leblanc, A.; Gibson, A.; Hong, K.; Kim, J.; Janke, L.; Li, L.; Vasilyeva, A.; Finkelstein, D.; Sprowl, J. Identification of OAT1/OAT3 as contributors to cisplatin toxicity. Clin. Transl. Sci. 2017, 10, 412–420.
- Morsy, M.A.; Heeba, G.H. Nebivolol ameliorates cisplatin-induced nephrotoxicity in rats. Basic Clin. Pharmacol. Toxicol. 2016, 118, 449–455.
- Nakamura, T.; Yonezawa, A.; Hashimoto, S.; Katsura, T.; Inui, K. Disruption of multidrug and toxin extrusion MATE1 potentiates cisplatin-induced nephrotoxicity. Biochem. Pharm. 2010, 80, 1762–1767.
- Wen, X.; Buckley, B.; McCandlish, E.; Goedken, M.J.; Syed, S.; Pelis, R.; Manautou, J.E.; Aleksunes, L.M. Transgenic expression of the human MRP2 transporter reduces cisplatin accumulation and nephrotoxicity in Mrp2-null mice. Am. J. Pathol. 2014, 184, 1299–1308.
- Townsend, D.M.; Deng, M.; Zhang, L.; Lapus, M.G.; Hanigan, M.H. Metabolism of cisplatin to a nephrotoxin in proximal tubule cells. J. Am. Soc. Nephrol. 2003, 14, 1–10.
- Nieskens, T.T.; Peters, J.G.; Dabaghie, D.; Korte, D.; Jansen, K.; Van Asbeck, A.H.; Tavraz, N.N.; Friedrich, T.; Russel, F.G.; Masereeuw, R. Expression of organic anion transporter 1 or 3 in human kidney proximal tubule cells reduces cisplatin sensitivity. Drug Metab. Dispos. 2018, 46, 592–599.
- Jhaveri, K.D.; Wanchoo, R.; Sakhiya, V.; Ross, D.W.; Fishbane, S. Adverse renal effects of novel molecular oncologic targeted therapies: A narrative review. Kidney Int. Rep. 2017, 2, 108–123.
- Marcolino, M.; Boersma, E.; Clementino, N.; Macedo, A.; Marx-Neto, A.; Silva, M.; van Gelder, T.; Akkerhuis, K.; Ribeiro, A. Imatinib treatment duration is related to decreased estimated glomerular filtration rate in chronic myeloid leukemia patients. Ann. Oncol. 2011, 22, 2073–2079.
- Huang, D.; Wang, C.; Duan, Y.; Meng, Q.; Liu, Z.; Huo, X.; Sun, H.; Ma, X.; Liu, K. Targeting Oct2 and P53: Formononetin prevents cisplatin-induced acute kidney injury. Toxicol. Appl. Pharmacol. 2017, 326, 15–24.
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302.
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Invest. 2011, 121, 4210–4221.
- Lin, X.; Okuda, T.; Holzer, A.; Howell, S. The Copper Transporter CTR1 Regulates Cisplatin Uptake in Saccharomyces cerevisiae. Mol. Pharmacol. 2002, 62, 1154–1159.
- Fox, E.; Levin, K.; Zhu, Y.; Segers, B.; Balamuth, N.; Womer, R.; Bagatell, R.; Balis, F. Pantoprazole, an Inhibitor of the Organic Cation Transporter 2, Does Not Ameliorate Cisplatin-Related Ototoxicity or Nephrotoxicity in Children and Adolescents with Newly Diagnosed Osteosarcoma Treated with Methotrexate, Doxorubicin, and Cisplatin. Oncologist 2018, 23, 762.
- Kuo, M.T.; Chen, H.H.; Song, I.S.; Savaraj, N.; Ishikawa, T. The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev. 2007, 26, 71–83.
- Hoffmann, E.K.; Sørensen, B.H.; Sauter, D.P.; Lambert, I.H. Role of volume-regulated and calcium-activated anion channels in cell volume homeostasis, cancer and drug resistance. Channels 2015, 9, 380–396.
- Osei-Owusu, J.; Yang, J.; Del Carmen Vitery, M.; Qiu, Z. Molecular biology and physiology of volume-regulated anion channel (VRAC). In Current Topics in Membranes; Elsevier: Amsterdam, The Netherlands, 2018; Volume 81, pp. 177–203.
- Holditch, S.J.; Brown, C.N.; Lombardi, A.M.; Nguyen, K.N.; Edelstein, C.L. Recent Advances in Models, Mechanisms, Biomarkers, and Interventions in Cisplatin-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2019, 20, 3011.
- Lalan, M.; Bagchi, T.; Misra, A. The Cell. In Challenges in Delivery of Therapeutic Genomics and Proteomics; Elsevier: Amsterdam, The Netherlands, 2011; pp. 1–43.
- Yonny, M.E.; García, E.M.; Lopez, A.; Arroquy, J.I.; Nazareno, M.A. Measurement of malondialdehyde as oxidative stress biomarker in goat plasma by HPLC-DAD. Microchem. J. 2016, 129, 281–285.
- Zhang, Y.; Chen, Y.; Li, B.; Ding, P.; Jin, D.; Hou, S.; Cai, X.; Sheng, X. The effect of monotropein on alleviating cisplatin-induced acute kidney injury by inhibiting oxidative damage, inflammation and apoptosis. Biomed. Pharmacother. 2020, 129, 110408.
- Tan, R.Z.; Wang, C.; Deng, C.; Zhong, X.; Yan, Y.; Luo, Y.; Lan, H.Y.; He, T.; Wang, L. Quercetin protects against cisplatin-induced acute kidney injury by inhibiting Mincle/Syk/NF-κB signaling maintained macrophage inflammation. Phytother. Res. 2020, 34, 139–152.
- Mukhopadhyay, P.; Horváth, B.; Kechrid, M.; Tanchian, G.; Rajesh, M.; Naura, A.S.; Boulares, A.H.; Pacher, P. Poly (ADP-ribose) polymerase-1 is a key mediator of cisplatin-induced kidney inflammation and injury. Free Radic. Biol. Med. 2011, 51, 1774–1788.
- Malik, S.; Suchal, K.; Gamad, N.; Dinda, A.K.; Arya, D.S.; Bhatia, J. Telmisartan ameliorates cisplatin-induced nephrotoxicity by inhibiting MAPK mediated inflammation and apoptosis. Eur. J. Pharm. 2015, 748, 54–60.
- Furuichi, K.; Kaneko, S.; Wada, T. Chemokine/chemokine receptor-mediated inflammation regulates pathologic changes from acute kidney injury to chronic kidney disease. Clin. Exp. Nephrol. 2009, 13, 9–14.
- Liu, X.-Q.; Jin, J.; Li, Z.; Jiang, L.; Dong, Y.-H.; Cai, Y.-T.; Wu, M.-F.; Wang, J.-N.; Ma, T.-T.; Wen, J.-G. Rutaecarpine Derivative Cpd-6c Alleviates Acute Kidney Injury by Targeting PDE4B, a Key Enzyme Mediating inflammation in Cisplatin Nephropathy. Biochem. Pharmacol. 2020, 180, 114132.
- Mitazaki, S.; Honma, S.; Suto, M.; Kato, N.; Hiraiwa, K.; Yoshida, M.; Abe, S. Interleukin-6 plays a protective role in development of cisplatin-induced acute renal failure through upregulation of anti-oxidative stress factors. Life Sci. 2011, 88, 1142–1148.
- Liu, M.; Chien, C.-C.; Burne-Taney, M.; Molls, R.R.; Racusen, L.C.; Colvin, R.B.; Rabb, H. A pathophysiologic role for T lymphocytes in murine acute cisplatin nephrotoxicity. J. Am. Soc. Nephrol. 2006, 17, 765–774.
- Gao, Z.; Liu, G.; Hu, Z.; Li, X.; Yang, X.; Jiang, B.; Li, X. Grape seed proanthocyanidin extract protects from cisplatin-induced nephrotoxicity by inhibiting endoplasmic reticulum stress-induced apoptosis. Mol. Med. Rep. 2014, 9, 801–807.
- Tan, Z.; Guo, F.; Huang, Z.; Xia, Z.; Liu, J.; Tao, S.; Li, L.; Feng, Y.; Du, X.; Ma, L. Pharmacological and genetic inhibition of fatty acid-binding protein 4 alleviated cisplatin-induced acute kidney injury. J. Cell. Mol. Med. 2019, 23, 6260–6270.
- Zarember, K.A.; Godowski, P.J. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 2002, 168, 554–561.
- Kimbrell, D.A.; Beutler, B. The evolution and genetics of innate immunity. Nat. Rev. Genet. 2001, 2, 256–267.
- Aderem, A.; Ulevitch, R.J. Toll-like receptors in the induction of the innate immune response. Nature 2000, 406, 782–787.
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801.
- Godfroy, J.I., III; Roostan, M.; Moroz, Y.S.; Korendovych, I.V.; Yin, H. Isolated Toll-like receptor transmembrane domains are capable of oligomerization. PloS ONE 2012, 7, e48875.
- Bell, J.K.; Mullen, G.E.; Leifer, C.A.; Mazzoni, A.; Davies, D.R.; Segal, D.M. Leucine-rich repeats and pathogen recognition in Toll-like receptors. Trends Immunol. 2003, 24, 528–533.
- Butcher, S.K.; O’Carroll, C.E.; Wells, C.A.; Carmody, R.J. Toll-like receptors drive specific patterns of tolerance and training on restimulation of macrophages. Front. Immunol. 2018, 9, 933.
- Zhang, D.; Zhang, G.; Hayden, M.S.; Greenblatt, M.B.; Bussey, C.; Flavell, R.A.; Ghosh, S. A toll-like receptor that prevents infection by uropathogenic bacteria. Science 2004, 303, 1522–1526.
- Sharma, S.; Zou, W.; Sun, Q.; Grandvaux, N.; Julkunen, I.; Hemmi, H.; Yamamoto, M.; Akira, S.; Yeh, W.-C.; Lin, R. Activation of TBK1 and IKKε kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J. Virol. 2004, 78, 10636–10649.
- Patel, S. Danger-associated molecular patterns (DAMPs): The derivatives and triggers of inflammation. Curr. Allergy Asthma Rep. 2018, 18, 63.
- Kurup, S.P.; Tarleton, R.L. Perpetual expression of PAMPs necessary for optimal immune control and clearance of a persistent pathogen. Nat. Commun. 2013, 4, 2616.
- Piccinini, A.; Midwood, K. DAMPening inflammation by modulating TLR signalling. Mediat. Inflamm. 2010, 2010, 672395.
- Bagchi, A.; Herrup, E.A.; Warren, H.S.; Trigilio, J.; Shin, H.-S.; Valentine, C.; Hellman, J. MyD88-dependent and MyD88-independent pathways in synergy, priming, and tolerance between TLR agonists. J. Immunol. 2007, 178, 1164–1171.
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643.
- Mäkelä, S.M.; Strengell, M.; Pietilä, T.E.; Österlund, P.; Julkunen, I. Multiple signaling pathways contribute to synergistic TLR ligand-dependent cytokine gene expression in human monocyte-derived macrophages and dendritic cells. J. Leukoc. Biol. 2009, 85, 664–672.
- Grassin-Delyle, S.; Abrial, C.; Salvator, H.; Brollo, M.; Naline, E.; Devillier, P. The role of toll-like receptors in the production of cytokines by human lung macrophages. J. Innate Immun. 2020, 12, 63–73.
- Wörnle, M.; Schmid, H.; Banas, B.; Merkle, M.; Henger, A.; Roeder, M.; Blattner, S.; Bock, E.; Kretzler, M.; Gröne, H.-J. Novel role of toll-like receptor 3 in hepatitis C-associated glomerulonephritis. Am. J. Pathol. 2006, 168, 370–385.
- Kropf, P.; Freudenberg, M.A.; Modolell, M.; Price, H.P.; Herath, S.; Antoniazi, S.; Galanos, C.; Smith, D.F.; Müller, I. Toll-like receptor 4 contributes to efficient control of infection with the protozoan parasite Leishmania major. Infect. Immun. 2004, 72, 1920–1928.
- Shigeoka, A.A.; Holscher, T.D.; King, A.J.; Hall, F.W.; Kiosses, W.B.; Tobias, P.S.; Mackman, N.; McKay, D.B. TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and-independent pathways. J. Immunol. 2007, 178, 6252–6258.
- Conti, F.; Spinelli, F.R.; Truglia, S.; Miranda, F.; Alessandri, C.; Ceccarelli, F.; Bombardieri, M.; Giannakakis, K.; Valesini, G. Kidney expression of toll like receptors in lupus nephritis: Quantification and clinicopathological correlations. Mediat. Inflamm. 2016, 2016.
- Ciferska, H.; Horak, P.; Konttinen, Y.T.; Krejci, K.; Tichy, T.; Hermanova, Z.; Zadrazil, J. Expression of nucleic acid binding Toll-like receptors in control, lupus and transplanted kidneys–a preliminary pilot study. Lupus 2008, 17, 580–585.
- Lin, M.; Yiu, W.H.; Wu, H.J.; Chan, L.Y.; Leung, J.C.; Au, W.S.; Chan, K.W.; Lai, K.N.; Tang, S.C. Toll-like receptor 4 promotes tubular inflammation in diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 86–102.
- Srivastava, T.; Sharma, M.; Yew, K.-H.; Sharma, R.; Duncan, R.S.; Saleem, M.A.; McCarthy, E.T.; Kats, A.; Cudmore, P.A.; Alon, U.S. LPS and PAN-induced podocyte injury in an in vitro model of minimal change disease: Changes in TLR profile. J. Cell Commun. Signal. 2013, 7, 49–60.
- Shimada, M.; Ishimoto, T.; Lee, P.Y.; Lanaspa, M.A.; Rivard, C.J.; Roncal-Jimenez, C.A.; Wymer, D.T.; Yamabe, H.; Mathieson, P.W.; Saleem, M.A. Toll-like receptor 3 ligands induce CD80 expression in human podocytes via an NF-κ B-dependent pathway. Nephrol. Dial. Transplant. 2012, 27, 81–89.
- Li, D.; Zou, L.; Feng, Y.; Xu, G.; Gong, Y.; Zhao, G.; Ouyang, W.; Thurman, J.M.; Chao, W. Complement factor B production in renal tubular cells and its role in sodium transporter expression during polymicrobial sepsis. Crit. Care Med. 2016, 44, e289.
- Bäckhed, F.; Söderhäll, M.; Ekman, P.; Normark, S.; Richter-Dahlfors, A. Induction of innate immune responses by Escherichia coli and purified lipopolysaccharide correlate with organ-and cell-specific expression of Toll-like receptors within the human urinary tract. Cell. Microbiol. 2001, 3, 153–158.
- Jonassen, J.A.; Kohjimoto, Y.; Scheid, C.R.; Schmidt, M. Oxalate toxicity in renal cells. Urol. Res. 2005, 33, 329–339.
- Yang, W.S.; Kim, J.-S.; Han, N.J.; Lee, M.J.; Park, S.-K. Toll-like receptor 4/spleen tyrosine kinase complex in high glucose signal transduction of proximal tubular epithelial cells. Cell. Physiol. Biochem. 2015, 35, 2309–2319.
- Han, S.J.; Li, H.; Kim, M.; Shlomchik, M.J.; Lee, H.T. Kidney proximal tubular TLR9 exacerbates ischemic acute kidney injury. J. Immunol. 2018, 201, 1073–1085.
- Samuelsson, P.; Hang, L.; Wullt, B.; Irjala, H.; Svanborg, C. Toll-like receptor 4 expression and cytokine responses in the human urinary tract mucosa. Infect. Immun. 2004, 72, 3179–3186.
- Papadimitraki, E.; Tzardi, M.; Bertsias, G.; Sotsiou, E.; Boumpas, D.T. Glomerular expression of toll-like receptor-9 in lupus nephritis but not in normal kidneys: Implications for the amplification of the inflammatory response. Lupus 2009, 18, 831–835.
- González-Guerrero, C.; Cannata-Ortiz, P.; Guerri, C.; Egido, J.; Ortiz, A.; Ramos, A.M. TLR4-mediated inflammation is a key pathogenic event leading to kidney damage and fibrosis in cyclosporine nephrotoxicity. Arch. Toxicol. 2017, 91, 1925–1939.
- Alikhan, M.A.; Summers, S.A.; Gan, P.Y.; Chan, A.J.; Khouri, M.B.; Ooi, J.D.; Ghali, J.R.; Odobasic, D.; Hickey, M.J.; Kitching, A.R. Endogenous toll-like receptor 9 regulates AKI by promoting regulatory T cell recruitment. J. Am. Soc. Nephrol. 2016, 27, 706–714.
- Lepenies, J.; Eardley, K.S.; Kienitz, T.; Hewison, M.; Ihl, T.; Stewart, P.M.; Cockwell, P.; Quinkler, M. Renal TLR4 mRNA expression correlates with inflammatory marker MCP-1 and profibrotic molecule TGF-β1 in patients with chronic kidney disease. Nephron Clin. Pract. 2011, 119, c97–c104.
- Leemans, J.C.; Stokman, G.; Claessen, N.; Rouschop, K.M.; Teske, G.J.; Kirschning, C.J.; Akira, S.; Van der Poll, T.; Weening, J.J.; Florquin, S. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J. Clin. Investig. 2005, 115, 2894–2903.
- Verzola, D.; Cappuccino, L.; D’amato, E.; Villaggio, B.; Gianiorio, F.; Mij, M.; Simonato, A.; Viazzi, F.; Salvidio, G.; Garibotto, G. Enhanced glomerular Toll-like receptor 4 expression and signaling in patients with type 2 diabetic nephropathy and microalbuminuria. Kidney Int. 2014, 86, 1229–1243.
- Andrade-Silva, M.; Cenedeze, M.A.; Perandini, L.A.; Felizardo, R.J.F.; Watanabe, I.K.M.; Agudelo, J.S.H.; Castoldi, A.; Gonçalves, G.M.; Origassa, C.S.T.; Semedo, P. TLR2 and TLR4 play opposite role in autophagy associated with cisplatin-induced acute kidney injury. Clin. Sci. 2018, 132, 1725–1739.
- Park, H.J.; Stokes, J.A.; Corr, M.; Yaksh, T.L. Toll-like receptor signaling regulates cisplatin-induced mechanical allodynia in mice. Cancer Chemother. Pharmacol. 2014, 73, 25–34.
- Volarevic, V.; Markovic, B.S.; Jankovic, M.G.; Djokovic, B.; Jovicic, N.; Harrell, C.R.; Fellabaum, C.; Djonov, V.; Arsenijevic, N.; Lukic, M.L. Galectin 3 protects from cisplatin-induced acute kidney injury by promoting TLR-2-dependent activation of IDO1/Kynurenine pathway in renal DCs. Theranostics 2019, 9, 5976.
- Ma, X.; Yan, L.; Zhu, Q.; Shao, F. Puerarin attenuates cisplatin-induced rat nephrotoxicity: The involvement of TLR4/NF-κB signaling pathway. PloS ONE 2017, 12, e0171612.
- Cenedeze, M.; Goncalves, G.; Feitoza, C.; Wang, P.; Damiao, M.; Bertocchi, A.; Pacheco-Silva, A.; Camara, N. The role of toll-like receptor 4 in cisplatin-induced renal injury. Transplant. Proc. 2007, 39, 409–411.
- Ramesh, G.; Zhang, B.; Uematsu, S.; Akira, S.; Reeves, W.B. Endotoxin and cisplatin synergistically induce renal dysfunction and cytokine production in mice. Am. J. Physiol. Ren. Physiol. 2007, 293, F325–F332.
- Babolmorad, G.; Latif, A.; Pollock, N.M.; Domingo, I.K.; Delyea, C.; Rieger, A.M.; Allison, W.T.; Bhavsar, A.P. Toll-like receptor 4 is activated by platinum and contributes to cisplatin-induced ototoxicity. bioRxiv 2020, e51280.
- Dessing, M.C.; Kers, J.; Damman, J.; Leuvenink, H.G.; Van Goor, H.; Hillebrands, J.-L.; Hepkema, B.G.; Snieder, H.; Van den Born, J.; De Borst, M.H. Toll-like receptor family polymorphisms are associated with primary renal diseases but not with renal outcomes following kidney transplantation. PloS ONE 2015, 10, e0139769.
- Elloumi, N.; Fakhfakh, R.; Abida, O.; Ayadi, L.; Marzouk, S.; Hachicha, H.; Fourati, M.; Bahloul, Z.; Mhiri, M.; Kammoun, K. Relevant genetic polymorphisms and kidney expression of Toll-like receptor (TLR)-5 and TLR-9 in lupus nephritis. Clin. Exp. Immunol. 2017, 190, 328–339.
- Ducloux, D.; Deschamps, M.; Yannaraki, M.; Ferrand, C.; Bamoulid, J.; Saas, P.; Kazory, A.; Chalopin, J.-M.; Tiberghien, P. Relevance of Toll-like receptor-4 polymorphisms in renal transplantation. Kidney Int. 2005, 67, 2454–2461.
- Santos-Martins, M.; Sameiro-Faria, M.; Ribeiro, S.; Rocha-Pereira, P.; Nascimento, H.; Reis, F.; Miranda, V.; Quintanilha, A.; Belo, L.; Beirão, I. TLR4 and TLR9 polymorphisms effect on inflammatory response in end-stage renal disease patients. Eur. J. Inflamm. 2014, 12, 521–529.
- Farhat, K.; Riekenberg, S.; Heine, H.; Debarry, J.; Lang, R.; Mages, J.; Buwitt-Beckmann, U.; Röschmann, K.; Jung, G.; Wiesmüller, K.H. Heterodimerization of TLR2 with TLR1 or TLR6 expands the ligand spectrum but does not lead to differential signaling. J. Leukoc. Biol. 2008, 83, 692–701.
- Guan, Y.; Ranoa, D.R.E.; Jiang, S.; Mutha, S.K.; Li, X.; Baudry, J.; Tapping, R.I. Human TLRs 10 and 1 share common mechanisms of innate immune sensing but not signaling. J. Immunol. 2010, 184, 5094–5103.
- Matsushima, N.; Tanaka, T.; Enkhbayar, P.; Mikami, T.; Taga, M.; Yamada, K.; Kuroki, Y. Comparative sequence analysis of leucine-rich repeats (LRRs) within vertebrate toll-like receptors. Bmc Genom. 2007, 8, 124.
- De Oliviera Nascimento, L.; Massari, P.; Wetzler, L.M. The role of TLR2 in infection and immunity. Front. Immunol. 2012, 3, 79.
- Raby, A.-C.; González-Mateo, G.T.; Williams, A.; Topley, N.; Fraser, D.; López-Cabrera, M.; Labéta, M.O. Targeting Toll-like receptors with soluble Toll-like receptor 2 prevents peritoneal dialysis solution–induced fibrosis. Kidney Int. 2018, 94, 346–362.
- Shen, Q.; Zhang, X.; Li, Q.; Zhang, J.; Lai, H.; Gan, H.; Du, X.; Li, M. TLR2 protects cisplatin-induced acute kidney injury associated with autophagy via PI3K/Akt signaling pathway. J. Cell. Biochem. 2019, 120, 4366–4374.
- Takahashi, A.; Kimura, T.; Takabatake, Y.; Namba, T.; Kaimori, J.; Kitamura, H.; Matsui, I.; Niimura, F.; Matsusaka, T.; Fujita, N. Autophagy guards against cisplatin-induced acute kidney injury. Am. J. Pathol. 2012, 180, 517–525.
- Myeku, N.; Figueiredo-Pereira, M.E. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy association With sequestosome 1/p62. J. Biol. Chem. 2011, 286, 22426–22440.
- Cherra III, S.J.; Kulich, S.M.; Uechi, G.; Balasubramani, M.; Mountzouris, J.; Day, B.W.; Chu, C.T. Regulation of the autophagy protein LC3 by phosphorylation. J. Cell Biol. 2010, 190, 533–539.
- Barth, S.; Glick, D.; Macleod, K.F. Autophagy: Assays and artifacts. J. Pathol. 2010, 221, 117–124.
- Xu, Y.; Chen, S.; Cao, Y.; Zhou, P.; Chen, Z.; Cheng, K. Discovery of novel small molecule TLR4 inhibitors as potent anti-inflammatory agents. Eur. J. Med. Chem. 2018, 154, 253–266.
- Zhang, B.; Ramesh, G.; Uematsu, S.; Akira, S.; Reeves, W.B. TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. J. Am. Soc. Nephrol. 2008, 19, 923–932.
- Park, B.S.; Lee, J.-O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, e66.
- Haynes, L.M.; Moore, D.D.; Kurt-Jones, E.A.; Finberg, R.W.; Anderson, L.J.; Tripp, R.A. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J. Virol. 2001, 75, 10730–10737.
- Kurt-Jones, E.A.; Popova, L.; Kwinn, L.; Haynes, L.M.; Jones, L.P.; Tripp, R.A.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000, 1, 398–401.
- Lai, C.-Y.; Strange, D.P.; Wong, T.A.S.; Lehrer, A.T.; Verma, S. Ebola virus glycoprotein induces an innate immune response in vivo via TLR4. Front. Microbiol. 2017, 8, 1571.
- Rashidi, N.; Mirahmadian, M.; Jeddi-Tehrani, M.; Rezania, S.; Ghasemi, J.; Kazemnejad, S.; Mirzadegan, E.; Vafaei, S.; Kashanian, M.; Rasoulzadeh, Z. Lipopolysaccharide-and lipoteichoic acid-mediated pro-inflammatory cytokine production and modulation of TLR2, TLR4 and MyD88 expression in human endometrial cells. J. Reprod. Infertil. 2015, 16, 72.
- Thomas, P.G.; Carter, M.R.; Atochina, O.; Da’Dara, A.A.; Piskorska, D.; McGuire, E.; Harn, D.A. Maturation of dendritic cell 2 phenotype by a helminth glycan uses a Toll-like receptor 4-dependent mechanism. J. Immunol. 2003, 171, 5837–5841.
- Oh, S.-M.; Park, G.; Lee, S.H.; Seo, C.-S.; Shin, H.-K.; Oh, D.-S. Assessing the recovery from prerenal and renal acute kidney injury after treatment with single herbal medicine via activity of the biomarkers HMGB1, NGAL and KIM-1 in kidney proximal tubular cells treated by cisplatin with different doses and exposure times. BMC Complementary Altern. Med. 2017, 17, 544.
- Ullah, M.; Liu, D.D.; Rai, S.; Concepcion, W.; Thakor, A.S. HSP70-Mediated NLRP3 Inflammasome Suppression Underlies Reversal of Acute Kidney Injury Following Extracellular Vesicle and Focused Ultrasound Combination Therapy. Int. J. Mol. Sci. 2020, 21, 4085.
- Rachmawati, D.; Bontkes, H.J.; Verstege, M.I.; Muris, J.; Von Blomberg, B.M.E.; Scheper, R.J.; Van Hoogstraten, I.M. Transition metal sensing by Toll-like receptor-4: Next to nickel, cobalt and palladium are potent human dendritic cell stimulators. Contact Dermat. 2013, 68, 331–338.
- Fenhammar, J.; Rundgren, M.; Hultenby, K.; Forestier, J.; Taavo, M.; Kenne, E.; Weitzberg, E.; Eriksson, S.; Ozenci, V.; Wernerson, A. Renal effects of treatment with a TLR4 inhibitor in conscious septic sheep. Crit. Care 2014, 18, 488.
- Fenhammar, J.; Rundgren, M.; Forestier, J.; Kalman, S.; Eriksson, S.; Frithiof, R. Toll-like receptor 4 inhibitor TAK-242 attenuates acute kidney injury in endotoxemic sheep. Anesthesiol. J. Am. Soc. Anesthesiol. 2011, 114, 1130–1137.
- Yamada, M.; Ichikawa, T.; Ii, M.; Sunamoto, M.; Itoh, K.; Tamura, N.; Kitazaki, T. Discovery of novel and potent small-molecule inhibitors of NO and cytokine production as antisepsis agents: Synthesis and biological activity of alkyl 6-(N-substituted sulfamoyl) cyclohex-1-ene-1-carboxylate. J. Med. Chem. 2005, 48, 7457–7467.
- Takashima, K.; Matsunaga, N.; Yoshimatsu, M.; Hazeki, K.; Kaisho, T.; Uekata, M.; Hazeki, O.; Akira, S.; Iizawa, Y.; Ii, M. Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Br. J. Pharmacol. 2009, 157, 1250–1262.
- Kashani, B.; Zandi, Z.; Karimzadeh, M.R.; Bashash, D.; Nasrollahzadeh, A.; Ghaffari, S.H. Blockade of TLR4 using TAK-242 (resatorvid) enhances anti-cancer effects of chemotherapeutic agents: A novel synergistic approach for breast and ovarian cancers. Immunol. Res. 2019, 67, 505–516.
- Pedersen, G.; Andresen, L.; Matthiessen, M.; Rask-Madsen, J.; Brynskov, J. Expression of Toll-like receptor 9 and response to bacterial CpG oligodeoxynucleotides in human intestinal epithelium. Clin. Exp. Immunol. 2005, 141, 298–306.
- Wang, Y.; Abel, K.; Lantz, K.; Krieg, A.M.; McChesney, M.B.; Miller, C.J. The Toll-like receptor 7 (TLR7) agonist, imiquimod, and the TLR9 agonist, CpG ODN, induce antiviral cytokines and chemokines but do not prevent vaginal transmission of simian immunodeficiency virus when applied intravaginally to rhesus macaques. J. Virol. 2005, 79, 14355–14370.
- Wu, X.; Peng, S.L. Toll-like receptor 9 signaling protects against murine lupus. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2006, 54, 336–342.
- Machida, H.; Ito, S.; Hirose, T.; Takeshita, F.; Oshiro, H.; Nakamura, T.; Mori, M.; Inayama, Y.; Yan, K.; Kobayashi, N. Expression of Toll-like receptor 9 in renal podocytes in childhood-onset active and inactive lupus nephritis. Nephrol. Dial. Transplant. 2010, 25, 2430–2537.
- Mallavia, B.; Liu, F.; Lefrançais, E.; Cleary, S.J.; Kwaan, N.; Tian, J.J.; Magnen, M.; Sayah, D.M.; Soong, A.; Chen, J. Mitochondrial DNA stimulates TLR9-dependent neutrophil extracellular trap formation in primary graft dysfunction. Am. J. Respir. Cell Mol. Biol. 2020, 62, 364–372.
- Busconi, L.; Bauer, J.W.; Tumang, J.R.; Laws, A.; Perkins-Mesires, K.; Tabor, A.S.; Lau, C.; Corley, R.B.; Rothstein, T.L.; Lund, F.E. Functional outcome of B cell activation by chromatin immune complex engagement of the B cell receptor and TLR9. J. Immunol. 2007, 179, 7397–7405.
- Brooks, J.; Sun, W.; Chiosis, G.; Leifer, C.A. Heat shock protein gp96 regulates Toll-like receptor 9 proteolytic processing and confrimational stability. Biochem. Biophy. Res. Commun. 2012, 421, 780–784.
- Jiang, S.; Chen, X. Expression of high-mobility group box 1 protein (HMGB1) and toll-like receptor 9 (TLR9) in retinas of diabetic rats. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2017, 23, 3115.
- Saito, K.; Kukita, K.; Kutomi, G.; Okuya, K.; Asanuma, H.; Tabeya, T.; Naishiro, Y.; Yamamoto, M.; Takahashi, H.; Torigoe, T. Heat shock protein 90 associates with Toll-like receptors 7/9 and mediates self-nucleic acid recognition in SLE. Eur. J. Immunol. 2015, 45, 2028–2041.
- Urry, Z.; Xystrakis, E.; Richards, D.F.; McDonald, J.; Sattar, Z.; Cousins, D.J.; Corrigan, C.J.; Hickman, E.; Brown, Z.; Hawrylowicz, C.M. Ligation of TLR9 induced on human IL-10–secreting Tregs by 1α, 25-dihydroxyvitamin D3 abrogates regulatory function. J. Clin. Investig. 2009, 119, 387–398.
- Lai, L.-W.; Yong, K.-C.; Lien, Y.-H.H. Pharmacologic recruitment of regulatory T cells as a therapy for ischemic acute kidney injury. Kidney Int. 2012, 81, 983–992.
- Humanes, B.; Camaño, S.; Lara, J.M.; Sabbisetti, V.; González-Nicolás, M.Á.; Bonventre, J.V.; Tejedor, A.; Lázaro, A. Cisplatin-induced renal inflammation is ameliorated by cilastatin nephroprotection. Nephrol. Dial. Transplant. 2017, 32, 1645–1655.
- Szlosarek, P.W.; Balkwill, F.R. Tumour necrosis factor α: A potential target for the therapy of solid tumours. Lancet Oncol. 2003, 4, 565–573.
- Wu, X.; Wu, M.-Y.; Jiang, M.; Zhi, Q.; Bian, X.; Xu, M.-D.; Gong, F.-R.; Hou, J.; Tao, M.; Shou, L.-M. TNF-α sensitizes chemotherapy and radiotherapy against breast cancer cells. Cancer Cell Int. 2017, 17, 13.
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? 1. Acta Pharmacol. Sin. 2008, 29, 1275–1288.
- Ozkok, A.; Ravichandran, K.; Wang, Q.; Ljubanovic, D.; Edelstein, C.L. NF-κB transcriptional inhibition ameliorates cisplatin-induced acute kidney injury (AKI). Toxicol. Lett. 2016, 240, 105–113.
- Ansari, M.A. Sinapic acid modulates Nrf2/HO-1 signaling pathway in cisplatin-induced nephrotoxicity in rats. Biomed. Pharmacother. 2017, 93, 646–653.
- Zhou, J.; An, C.; Jin, X.; Hu, Z.; Safirstein, R.L.; Wang, Y. TAK1 deficiency attenuates cisplatin-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 2020, 318, F209–F215.
- Zhou, J.; Fan, Y.; Zhong, J.; Huang, Z.; Huang, T.; Lin, S.; Chen, H. TAK1 mediates excessive autophagy via p38 and ERK in cisplatin-induced acute kidney injury. J. Cell. Mol. Med. 2018, 22, 2908–2921.
- Mihaly, S.; Ninomiya-Tsuji, J.; Morioka, S. TAK1 control of cell death. Cell Death Differ. 2014, 21, 1667–1676.
- Faubel, S.; Lewis, E.C.; Reznikov, L.; Ljubanovic, D.; Hoke, T.S.; Somerset, H.; Oh, D.-J.; Lu, L.; Klein, C.L.; Dinarello, C.A. Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1β, IL-18, IL-6, and neutrophil infiltration in the kidney. J. Pharmacol. Exp. Ther. 2007, 322, 8–15.
- Deng, J.; Kohda, Y.; Chiao, H.; Wang, Y.; Hu, X.; Hewitt, S.M.; Miyaji, T.; Mcleroy, P.; Nibhanupudy, B.; Li, S. Interleukin-10 inhibits ischemic and cisplatin-induced acute renal injury. Kidney Int. 2001, 60, 2118–2128.
- Lu, L.H.; Oh, D.-J.; Dursun, B.; He, Z.; Hoke, T.S.; Faubel, S.; Edelstein, C.L. Increased macrophage infiltration and fractalkine expression in cisplatin-induced acute renal failure in mice. J. Pharmacol. Exp. Ther. 2008, 324, 111–117.
- Tadagavadi, R.K.; Reeves, W.B. Endogenous IL-10 attenuates cisplatin nephrotoxicity: Role of dendritic cells. J. Immunol. 2010, 185, 4904–4911.
- Dhande, I.; Ma, W.; Hussain, T. Angiotensin AT 2 receptor stimulation is anti-inflammatory in lipopolysaccharide-activated THP-1 macrophages via increased interleukin-10 production. Hypertens. Res. 2015, 38, 21–29.
- Jones, V.S.; Huang, R.-Y.; Chen, L.-P.; Chen, Z.-S.; Fu, L.; Huang, R.-P. Cytokines in cancer drug resistance: Cues to new therapeutic strategies. Biochim. Biophys. Acta Rev. Cancer 2016, 1865, 255–265.
- Ravichandran, K.; Holditch, S.; Brown, C.N.; Wang, Q.; Ozkok, A.; Weiser-Evans, M.C.; Nemenoff, R.; Miyazaki, M.; Thiessen-Philbrook, H.; Parikh, C.R. IL-33 deficiency slows cancer growth but does not protect against cisplatin-induced AKI in mice with cancer. Am. J. Physiol. Ren. Physiol. 2018, 314, F356–F366.
- Akcay, A.; Nguyen, Q.; He, Z.; Turkmen, K.; Lee, D.W.; Hernando, A.A.; Altmann, C.; Toker, A.; Pacic, A.; Ljubanovic, D.G. IL-33 exacerbates acute kidney injury. J. Am. Soc. Nephrol. 2011, 22, 2057–2067.
- Scott, H.; Jones, D.; Pui, C. The tumor lysis syndrome. N. Engl. J. Med. 2011, 364, 1844–1854.
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro-and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 878–888.
- Dennen, P.; Altmann, C.; Kaufman, J.; Klein, C.L.; Andres-Hernando, A.; Ahuja, N.H.; Edelstein, C.L.; Cadnapaphornchai, M.A.; Keniston, A.; Faubel, S. Urine interleukin-6 is an early biomarker of acute kidney injury in children undergoing cardiac surgery. Crit. Care 2010, 14, 1–13.
- Mitazaki, S.; Kato, N.; Suto, M.; Hiraiwa, K.; Abe, S. Interleukin-6 deficiency accelerates cisplatin-induced acute renal failure but not systemic injury. Toxicology 2009, 265, 115–121.
- Suchi, K.; Fujiwara, H.; Okamura, S.; Okamura, H.; Umehara, S.; Todo, M.; Furutani, A.; Yoneda, M.; Shiozaki, A.; Kubota, T. Overexpression of Interleukin-6 suppresses cisplatin-induced cytotoxicity in esophageal squamous cell carcinoma cells. Anticancer Res. 2011, 31, 67–75.
- Reeves, W.B. Innate Immunity in Nephrotoxic Acute Kidney Injury. Trans. Am. Clin. Climatol. Assoc. 2019, 130, 33.
- Mollica Poeta, V.; Massara, M.; Capucetti, A.; Bonecchi, R. Chemokines and chemokine receptors: New targets for cancer immunotherapy. Front. Immunol. 2019, 10, 379.
- Liang, H.; Zhang, Z.; He, L.; Wang, Y. CXCL16 regulates cisplatin-induced acute kidney injury. Oncotarget 2016, 7, 31652.
- Hu, Z.B.; Chen, Y.; Gong, Y.X.; Gao, M.; Zhang, Y.; Wang, G.H.; Tang, R.N.; Liu, H.; Liu, B.C.; Ma, K.L. Activation of the CXCL16/CXCR6 pathway by inflammation contributes to atherosclerosis in patients with end-stage renal disease. Int. J. Med. Sci. 2016, 13, 858.
- Wehr, A.; Tacke, F. The roles of CXCL16 and CXCR6 in liver inflammation and fibrosis. Curr. Pathobiol. Rep. 2015, 3, 283–290.
- Lehrke, M.; Millington, S.C.; Lefterova, M.; Cumaranatunge, R.G.; Szapary, P.; Wilensky, R.; Rader, D.J.; Lazar, M.A.; Reilly, M.P. CXCL16 is a marker of inflammation, atherosclerosis, and acute coronary syndromes in humans. J. Am. Coll. Cardiol. 2007, 49, 442–449.
- Baeck, C.; Wehr, A.; Karlmark, K.R.; Heymann, F.; Vucur, M.; Gassler, N.; Huss, S.; Klussmann, S.; Eulberg, D.; Luedde, T. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012, 61, 416–426.
- Garcia, G.E.; Truong, L.D.; Li, P.; Zhang, P.; Johnson, R.J.; Wilson, C.B.; Feng, L. Inhibition of CXCL16 attenuates inflammatory and progressive phases of anti-glomerular basement membrane antibody-associated glomerulonephritis. Am. J. Pathol. 2007, 170, 1485–1496.
- Liang, H.; Ma, Z.; Peng, H.; He, L.; Hu, Z.; Wang, Y. CXCL16 deficiency attenuates renal injury and fibrosis in salt-sensitive hypertension. Sci. Rep. 2016, 6, 28715.
- Wehr, A.; Baeck, C.; Ulmer, F.; Gassler, N.; Hittatiya, K.; Luedde, T.; Neumann, U.P.; Trautwein, C.; Tacke, F. Pharmacological inhibition of the chemokine CXCL16 diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. PloS ONE 2014, 9, e112327.
- Sawant, K.V.; Poluri, K.M.; Dutta, A.K.; Sepuru, K.M.; Troshkina, A.; Garofalo, R.P.; Rajarathnam, K. Chemokine CXCL1 mediated neutrophil recruitment: Role of glycosaminoglycan interactions. Sci. Rep. 2016, 6, 33123.
- Ravindran, A.; Sawant, K.V.; Sarmiento, J.; Navarro, J.; Rajarathnam, K. Chemokine CXCL1 dimer is a potent agonist for the CXCR2 receptor. J. Biol. Chem. 2013, 288, 12244–12252.
- Kinsey, G.R.; Okusa, M.D. Role of leukocytes in the pathogenesis of acute kidney injury. Crit. Care 2012, 16, 1–5.