Podospora anserina is a filamentous fungus that, in contrast to most other fungi, is characterized by a defined limited lifespan. Already in the 1950s it was reported that this ascomycete develops a well-defined senescence syndrome. Depending on the strain, this syndrome occurs after a defined short period of growth (e.g., after 2–3 weeks): the pigmentation of the peripheral part of the thallus increases while the growth rate decreases until it comes to a complete stop and the thallus dies at the growth front. The molecular basis of aging of P. anserina have been carefully investigated over more than 60 years of research and a network of pathways and their interactions have been uncovered.
- aging
- autophagy
- homeostasis
- mitochondria
- peroxisomes
- Podospora anserina
- quality control
- signaling
1. Introduction
Subsequently, this phenotype was carefully investigated and it turned out to be under the control of environmental and genetic factors. Both nuclear as well as extranuclear genetic traits are active [1][2][3]. Later on, it was demonstrated that a genetic element located in mitochondria accumulates as a plasmid-like covalently closed circular DNA (plDNA). It behaves like a mobile element and gives rise to gross reorganization of the standard mitochondrial DNA (mtDNA). As a consequence, large parts of the mtDNA with a number of essential genes are deleted leading to deficiencies in mitochondrial biogenesis and function and death of the thallus at the hyphal tips [4][5][6][7][8].
Since this time, senescence in P. anserina was carefully analyzed and the fungus became a well-established model system in experimental aging research [9][10][11]. In particular, the analyses of a number of mutants, which live longer than the wild type, provided important clues and revealed insights into the mechanisms of lifespan control. This work unraveled a paramount role of mitochondria and of the cellular energy metabolism. One group of mutants (ex and mex) contained deletions of parts of the PaCoxI gene and, thus, an essential component of complex IV of the respiratory chain is ablated. In these mutants the expression of a nuclear gene coding for an alternative oxidase (PaAOX) is induced and respiratory deficiency is rescued [12][13]. As a consequence, the corresponding mutants are long-lived. The molecular basis of this example of mitochondrial-nuclear interactions, which requires signaling from impaired mitochondria to the nucleus and the activation of PaAox-specific transcription factors [14], became clear via the analysis of other mutants and uncovered an impact of ROS (for more details see below).
Subsequently, a number of different molecular pathways, involved in the control of cellular homeostasis, were identified which are effective in keeping the individual thallus functional over a longer period of time. However, when impairments accumulate beyond rescue limits, programmed cell death (PCD) [15] is induced and the thallus dies at the hyphal tips. PCD was found to be controlled by various factors like “apoptosis inducing factors” (AIFs) [16] and the activation of the two calcium-dependent metacaspases PaMCA1 and PaMCA2 [17][18][19]. During this process, the opening of a mitochondria transition pore (mPTP) plays a key role [20][21][22].
2. Biogenesis and Dynamics of Mitochondria
Mitochondria are semiautonomous organelles in which most of the approximately 1200–1600 proteins are encoded by nuclear DNA and only a few by mtDNA. Mitochondria are dynamic and change their morphology and ultrastructure depending on physiological constraints. Mitochondrial mass (size and number of mitochondria) changes during growth and development (Figure 1). This process is not the result of de novo synthesis of the organelle but by the biosynthesis of new components and their integration into existing mitochondria.
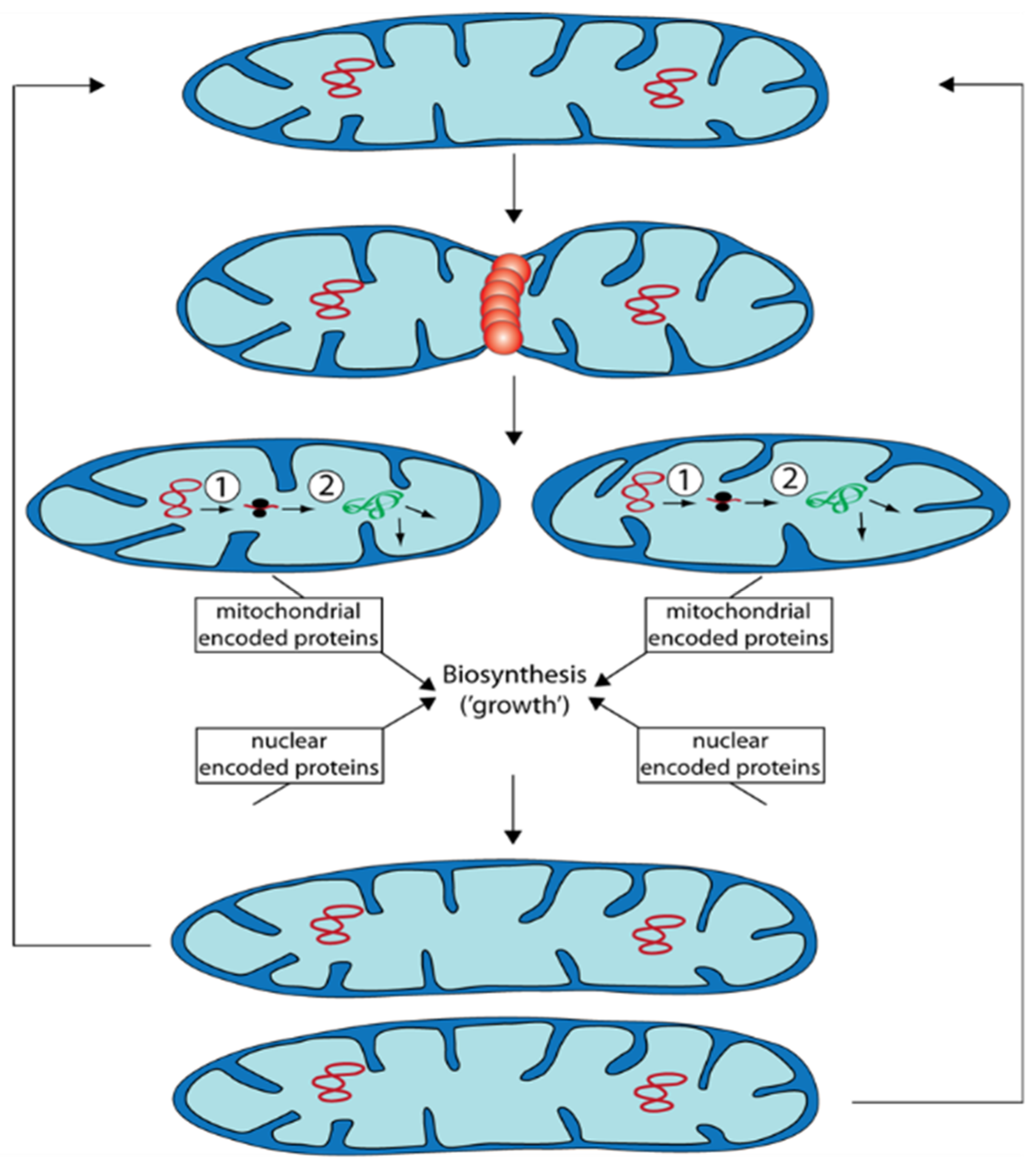
During “growth” of mitochondrial units, they form filamentous morphotypes that subsequently can divide into smaller units. These can fuse again to form filamentous organelles. Fission and fusion are genetically controlled by a number of proteins. Additionally, excess or functionally impaired (damaged) mitochondria can be removed by autophagy.
In addition to the processes of mitochondrial quality control discussed above the control of mitochondrial dynamics was demonstrated in P. anserina to have an effect on aging. Deletion of a gene coding for the dynamin-like protein PaDNM1, an essential protein involved in fission of mitochondria, led to an 11-fold increase in mean lifespan. Mitochondria of this strain had an strongly elongated morphology and even formed networks of fused filaments [23]. Only in very old cultures, mitochondria were found to be fragmented. Furthermore, in this strain no signs of typical reorganization of mtDNA found in the wild type occurred and the release of hydrogen peroxide was delayed to very old age. Lifespan extension was linked to an increase in resistance to the induction of programmed cell death. The relevance of PaDnm1 for normal aging of the wild type is indicated by the increased transcription of the gene in old cultures [23]. Computational modeling integrating mitochondrial fission and fusion, ROS stress, and mitophagy revealed a positive impact of mitochondrial dynamics in situations when mitochondria are only marginally damaged. In contrast, deceleration of fission and fusion is an advantage to reach a long lifespan when damage of mitochondria passed critical limits [24][25].
3. Perspectives: Potential Role of Peroxisomes
In a previous study, investigating the role of PaATG24, it was found that deletion of PaAtg24 reduces bulk autophagy, mitophagy and pexophagy [26]. Moreover, the number of peroxisomes, which slightly increase during wild-type aging, increases strongly in the short-lived PaAtg24 deletion mutant suggesting a role of peroxisomes in the aging process in P. anserina (Figure 2).
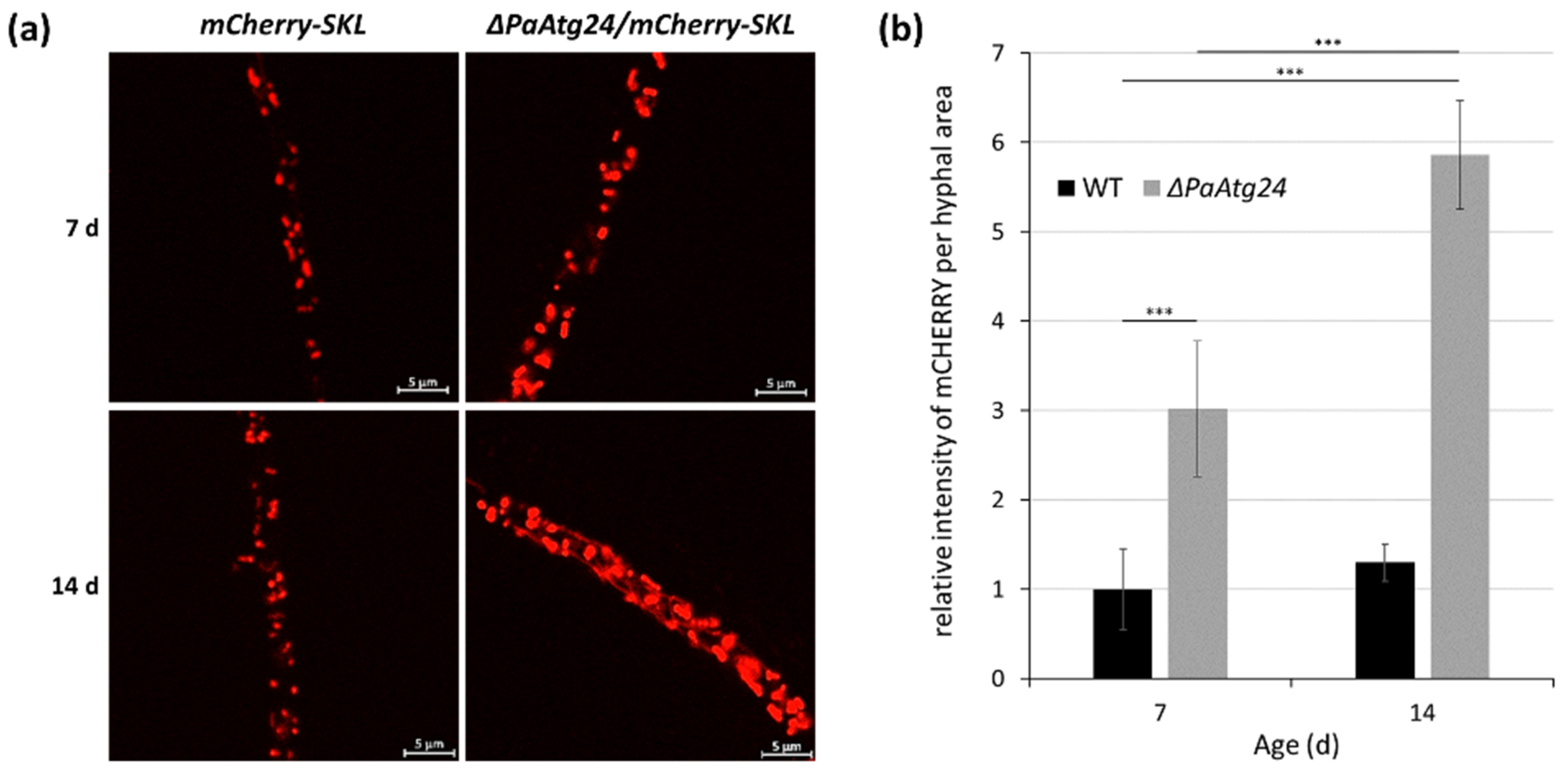
A key function of peroxisomes is to degrade fatty acids via ß-oxidation leading to the formation of acetyl-CoA that finally is further metabolized (i.e., in the Krebs cycle in mitochondria). In most organisms, from yeast to humans, during the first step of this process acyl-CoA oxidase performed the oxidation of the fatty acid, which leads to trans-Δ2-enoyl-CoA and the formation of hydrogen peroxide as a byproduct. This molecule contributes to the cellular ROS load, to ROS scavenging, signaling, and via unbalanced conditions to molecular damaging.
There is considerable evidence that a disturbance in peroxisomal redox homeostasis affects mitochondrial function and redox balance. For instance, the inactivation of peroxisomal catalase (for more details see above) in human cells results in functionally impaired mitochondria, which lose their ability to maintain a membrane potential and synthesize ROS themselves [27]. On the other hand, it was shown that enhancing the activity of peroxisomal catalase has beneficial effects on mitochondria. It is described that during aging peroxisomal protein import of peroxisomal catalase is becoming particularly impaired [28]. Enhancing the effectivity of catalase import into the peroxisomes, reduces cellular hydrogen peroxide levels, as well as the number of senescent cells in a population, and reverses mitochondrial depolarization [29].
This entry is adapted from the peer-reviewed paper 10.3390/jof7040263
References
- Esser, K.; Keller, W. Genes inhibiting senescence in the ascomycete Podospora anserina. Mol. Gen. Genet. 1976, 144, 107–110.
- Marcou, D. Notion de Longévité et Nature Cytoplasmique du Déterminant de la Sénescence chez Quelques Champignons; Masson: Paris, France, 1962; pp. 653–764.
- Marcou, D.; Schecroun, J. La sénescence chez Podospora pourrait etre due à des particules cytoplasmiques infectantes. C. R. Hebd. Seances Acad. Sci. 1959, 248, 280–283.
- Cummings, D.J.; Belcour, L.; Grandchamp, C. Mitochondrial DNA from Podospora anserina. II. Properties of mutant DNA and multimeric circular DNA from senescent cultures. Mol. Gen. Genet. 1979, 171, 239–250.
- Kück, U.; Osiewacz, H.D.; Schmidt, U.; Kappelhoff, B.; Schulte, E.; Stahl, U.; Esser, K. The onset of senescence is affected by DNA rearrangements of a discontinuous mitochondrial gene in Podospora anserina. Curr. Genet. 1985, 9, 373–382.
- Kück, U.; Stahl, U.; Esser, K. Plasmid-like DNA is part of mitochondrial DNA in Podospora anserina. Curr. Genet. 1981, 3, 151–156.
- Osiewacz, H.D.; Esser, K. The mitochondrial plasmid of Podospora anserina: A mobile intron of a mitochondrial gene. Curr. Genet. 1984, 8, 299–305.
- Stahl, U.; Lemke, P.A.; Tudzynski, P.; Kück, U.; Esser, K. Evidence for plasmid like DNA in a filamentous fungus, the ascomycete Podospora anserina. Mol. Gen. Genet. 1978, 162, 341–343.
- Osiewacz, H.D.; Hamann, A.; Zintel, S. Assessing organismal aging in the filamentous fungus Podospora anserina. Methods Mol. Biol. 2013, 965, 439–462.
- Osiewacz, H.D.; Stumpferl, S.W. Metabolism and aging in the filamentous fungus Podospora anserina. Arch. Gerontol. Geriatr. 2001, 32, 185–197.
- Scheckhuber, C.Q.; Osiewacz, H.D. Podospora anserina: A model organism to study mechanisms of healthy ageing. Mol. Genet. Genom. 2008, 280, 365–374.
- Belcour, L.; Begel, O.; Keller, A.M.; Vierny, C. Does senescence in Podospora anserina result from instability of the mitochondrial genome? In Mitochondrial Genes; Slonimski, P., Borst, P., Attardi, G., Eds.; Cold Spring Harbor: New York, NY, USA, 1982; pp. 415–421.
- Schulte, E.; Kück, U.; Esser, K. Extrachromosomal mutants from Podospora anserina: Permanent vegetative growth in spite of multiple recombination events in the mitochondrial genome. Mol. Gen. Genet. 1988, 211, 342–349.
- Sellem, C.H.; Bovier, E.; Lorin, S.; Sainsard-Chanet, A. Mutations in two zinc-cluster proteins activate alternative respiratory and gluconeogenic pathways and restore senescence in long-lived respiratory mutants of Podospora anserina. Genetics 2009, 182, 69–78.
- Carmona-Gutierrez, D.; Bauer, M.A.; Zimmermann, A.; Aguilera, A.; Austriaco, N.; Ayscough, K.; Balzan, R.; Bar-Nun, S.; Barrientos, A.; Belenky, P.; et al. Guidelines and recommendations on yeast cell death nomenclature. Microb. Cell 2018, 5, 4–31.
- Brust, D.; Hamann, A.; Osiewacz, H.D. Deletion of PaAif2 and PaAmid2, two genes encoding mitochondrial AIF-like oxidoreductases of Podospora anserina, leads to increased stress tolerance and lifespan extension. Curr. Genet. 2010, 56, 225–235.
- Hamann, A.; Brust, D.; Osiewacz, H.D. Deletion of putative apoptosis factors leads to lifespan extension in the fungal ageing model Podospora anserina. Mol. Microbiol. 2007, 65, 948–958.
- Minina, E.A.; Staal, J.; Alvarez, V.E.; Berges, J.A.; Berman-Frank, I.; Beyaert, R.; Bidle, K.D.; Bornancin, F.; Casanova, M.; Cazzulo, J.J.; et al. Classification and nomenclature of metacaspases and paracaspases: No more confusion with caspases. Mol. Cell 2020, 77, 927–929.
- Strobel, I.; Osiewacz, H.D. Poly(ADP-ribose) polymerase is a substrate recognized by two metacaspases of Podospora anserina. Eukaryot. Cell 2013, 12, 900–912.
- Brust, D.; Daum, B.; Breunig, C.; Hamann, A.; Kühlbrandt, W.; Osiewacz, H.D. Cyclophilin D links programmed cell death and organismal aging in Podospora anserina. Aging Cell 2010, 9, 761–775.
- Groebe, K.; Krause, F.; Kunstmann, B.; Unterluggauer, H.; Reifschneider, N.H.; Scheckhuber, C.Q.; Sastri, C.; Stegmann, W.; Wozny, W.; Schwall, G.P.; et al. Differential proteomic profiling of mitochondria from Podospora anserina, rat and human reveals distinct patterns of age-related oxidative changes. Exp. Gerontol. 2007, 42, 887–898.
- Kramer, P.; Jung, A.T.; Hamann, A.; Osiewacz, H.D. Cyclophilin D is involved in the regulation of autophagy and affects the lifespan of P. anserina in response to mitochondrial oxidative stress. Front. Genet. 2016, 7, 165.
- Scheckhuber, C.Q.; Erjavec, N.; Tinazli, A.; Hamann, A.; Nyström, T.; Osiewacz, H.D. Reducing mitochondrial fission results in increased life span and fitness of two fungal ageing models. Nat. Cell Biol. 2007, 9, 99–105.
- Figge, M.T.; Reichert, A.S.; Meyer-Hermann, M.; Osiewacz, H.D. Deceleration of fusion-fission cycles improves mitochondrial quality control during aging. PLoS Comput. Biol. 2012, 8, e1002576.
- Figge, M.T.; Osiewacz, H.D.; Reichert, A.S. Quality control of mitochondria during aging: Is there a good and a bad side of mitochondrial dynamics? Bioessays 2013, 35, 314–322.
- Henkel, V.; Schürmanns, L.; Brunner, M.; Hamann, A.; Osiewacz, H.D. Role of sorting nexin PaATG24 in autophagy, aging and development of Podospora anserina. Mech. Ageing Dev. 2020, 186, 111211.
- Koepke, J.I.; Wood, C.S.; Terlecky, L.J.; Walton, P.A.; Terlecky, S.R. Progeric effects of catalase inactivation in human cells. Toxicol. Appl. Pharmacol. 2008, 232, 99–108.
- Legakis, J.E.; Koepke, J.I.; Jedeszko, C.; Barlaskar, F.; Terlecky, L.J.; Edwards, H.J.; Walton, P.A.; Terlecky, S.R. Peroxisome senescence in human fibroblasts. Mol. Biol. Cell 2002, 13, 4243–4255.
- Koepke, J.I.; Nakrieko, K.A.; Wood, C.S.; Boucher, K.K.; Terlecky, L.J.; Walton, P.A.; Terlecky, S.R. Restoration of peroxisomal catalase import in a model of human cellular aging. Traffic 2007, 8, 1590–1600.