Regardless of the tumor evolution that is causing ITH, the existence of multiple distinctive cell populations in the same tumor has strong clinical implications [
48]. The diagnosis of primary and metastatic tumors is typically based on a single biopsy representing only a snapshot of ongoing tumor evolution and may be compromised by ITH [
49]. The studies described above show that a sole biopsy does not accurately capture the tumor’s genetic and phenotypic heterogeneity [
28,
46]. Therefore, multimodal strategies have to be taken into account to ensure patients receive effective and targeted therapy. In addition, it is necessary to develop new tools to study heterogeneity, and to identify new biomarkers of tumor heterogeneity. Further, besides focusing on clonal heterogeneity, nonclonal phenotypic heterogeneity should be taken into consideration, i.e., the fact that some cells respond to broad, environmental perturbations and drug treatments by conversion to many other cell states, including stem-like, resistant cell phenotypes [
50]. Finally, suitable models are needed that also take the effect of the tumor microenvironment into account.
3.2. Suitable In Vitro Strategies for Modeling Intra-Tumor Heterogeneity
Utilizing cancer models may complement or even supplement the approaches described above. ITH, the cellular interactions and the tumor’s molecular alterations could be investigated in cancer models derived from patients’ samples to study the underlying mechanisms and/or learn how ITH may direct clinical decision making.
Cancer models are naturally existing or artificially induced systems that share characteristics with human cancers [
85]. Experimental systems for the study of human cancer include genetically engineered mouse models (GEMMs) [
86,
87,
88,
89], two-dimensional (2D) cell lines [
90], patient-derived organoids (PDO) [
91,
92,
93,
94,
95], and patient-derived xenografts (PDX) [
95,
96] to study biochemical or genetic pathways and pathology of cancer. These in vivo and in vitro cancer models have been invaluable for our current understanding of cancer development and progression, as well as for therapy development. Further, these models are moving into focus regarding their potential use in cancer precision and/or personalized medicine [
97,
98].
Given the complexity and heterogeneity of cancer, a crucial question to be asked is whether these models are feasible to capture and investigate ITH.
GEMMs are created by inducing specific mutations in oncogenes and/or tumor suppressor genes and can be used to monitor tumorigenesis in vivo, but are limited by species differences in oncogenic pathogenesis, the shorter lifespan of mice, and often by the artificial simultaneous introduction of several oncogenic driver events [
88,
89].
Traditional 2D cell lines grow as monolayer, cultured on flat and rigid substrates [
91]. They have the advantage that they have once been derived from a cancer patient and are easier to manipulate in the laboratory, but they cannot completely replicate the environment of the patient tumor. Even within the same cancer model, data between laboratories are often irreproducible [
99,
100]. Nevertheless, cancer 2D cell lines have been used not only for in vitro but also for in vivo experiments, for example to generate xenograft models by subcutaneous injection of cancer cell lines into immunodeficient mice [
101].
Patient-derived xenografts (PDX) are generated by implantation of cancerous tissue from a patient’s tumor either under the skin (ectopic) or into the organ of tumor origin (orthotopic) and are most commonly used for preclinical drug development [
102,
103,
104].
Physiologically, cells grow in three dimensions (3D) to form discrete tissue and organ structures [
105,
106]. PDO models, growing in 3D, have been shown to reliably recapitulate the architecture of the donor tissue and to preserve its genomic background, therefore providing a highly relevant physiological system [
107]. With their optimal conditions for cellular proliferation, differentiation, and responsiveness to chemo- and targeted therapeutics, they recapitulate the functional tumor phenotype, including its ITH [
41].
The impact of adding a third culture dimension on the cellular drug response has been shown by Koch et al. [
108]. They compared the response of 2D colorectal cancer cell lines and 3D CRC cell line-derived spheroids to irradiation and chemotherapy [
108]. 3D CRC cell line cultures were more resistant to irradiation and chemotherapy, such as 5-FU and cisplatin, than their 2D counterparts [
108]. This must be taken into account when translating the results into clinical setting. Therefore, PDOs are increasingly used for studying tumor biology and the effects of targeted therapies [
109].
Schütte et al. investigated a colorectal cancer biobank comprising PDX models and PDO models, which were treated with clinically relevant compounds [
95]. They showed that PDOs recapitulate many of the genetic and transcriptomic features of the donor tumors whereas clonal discordances found at early passages were attributed to ITH [
95]. Ben-David et al. showed that the genomic and transcriptomic heterogeneity of cell lines impairs our ability to evaluate new therapeutics [
110]. Their results support efforts to systematically develop PDO models to reduce the reliance on poorly defined cell lines that were established before the next generation sequencing era [
110]. Further, Vlachogiannis et al. showed that PDOs can recapitulate patient responses in the clinic and could therefore be implemented as models for personalized oncology [
111].
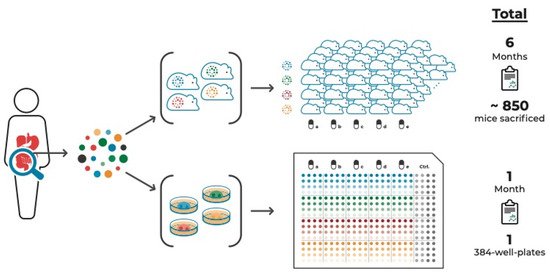
An additional advantage of PDO cultures is that they can be used in high-throughput drug screens. Such screens can be composed of multiple samples from the same tumor, thereby taking ITH into account [
41,
94]. compares this approach with a PDX approach aiming to test the same number of tumor samples and compounds to illustrate the time- and cost-effectiveness of the organoid system.
Figure 1. PDO vs. PDX cancer models to study a tumor’s drug response incorporating intra-tumor heterogeneity. Multi-regional sampling of a tumor is required to take ITH into account when assessing the tumor’s drug response. Compared to multi-regional PDX models, multi-regional PDO-based pre-therapeutic drug screenings are significantly more time- and cost-effective. These PDO-based screens, in addition to amplicon sequencing of multiple areas of tumor tissue and derived cell culture models, combined with targeted proteomic approaches, are both feasible and available within a timeframe that allows discussing guided treatment options.
Nevertheless, PDO are an in vitro culture system, therefore lacking, for example, liver or kidney clearance or liver pro-drug activation mechanism. Further, only the effect of the drug on the tumor (i.e., tumor-derived organoid) itself can be assessed, omitting potential toxicity on other organs or indirect effects asserted on, e.g., vasculature or hormone production in the pituitary gland. These aspects need to be taken into consideration for experimental design.
It is known that the microenvironment with tumor-surrounding and infiltrating cells, including fibroblasts and immune cells, have a major impact on drug response [
112,
113]. Another significant advantage of the organoid model system is the ability to study the interaction of cancer organoids with other specific cell types that can be introduced into a direct or indirect co-culture system. Indirect co-culture systems are based on the use of cell conditioned media. They are simple to apply and are therefore often used for in vitro experiments [
114]. However, they are not suitable for investigating the effects of cell contacts between cancer cells and stroma [
115]. Direct co-culture models are a closer representation of the in vivo scenario. Since the tumor microenvironment plays a critical role in tumorigenesis, 3D co-culture systems are used, including not only cancer cells but also stromal cells [
115]. Cancer cells were localized in a defined area within a stromal cell matrix to study the cytotoxic effect of anticancer drugs on both tumor and normal cells in the same system [
116].
Since antibodies against immune checkpoint proteins/receptors have shown clear clinical benefit for patients with advanced cancer, including melanoma, non-small cell lung cancer (NSCLC), and mismatch repair deficient (dMMR) colorectal cancer, organoid co-culture systems including immune cells are moving into the spotlight of current in vitro application [
117,
118,
119,
120,
121,
122,
123,
124,
125,
126]. Dijkstra et al. established a co-culture system of autologous tumor organoids and peripheral blood lymphocytes of patients to induce and analyze tumor-specific T cell responses for mismatch repair deficient colorectal cancer and non-small cell lung cancer in a personalized manner [
118]. Klein et al. demonstrated the advantages of co-culture systems of GBM organoids and human immune cells, to investigate not only immune–tumor interactions, but also to explore current and novel immunotherapies, such as adoptive T cell transfer, immune checkpoint inhibitors, or oncolytic viruses [
127].
Another promising technology for studying ITH is Organ-on-a-Chip (OoC), a culture model to mimic complex and dynamic in vivo microenvironments [
128]. An OoC is a multi-channel 3D microfluidic biochip, which recapitulates the activity, mechanism, and pathophysiological reaction of single-organ and multi-organ systems [
129]. It is a useful tool in controlling spatial arrangement of cell growth and fluids within micrometer-sized channels, which may be used to increase the physiological relevance of tumor models [
130]. OoC technology is expected to offer effective solutions to investigate the effects of drugs, as well as the causes of diseases and personalized therapeutic treatments [
131,
132,
133].
The development of a multi-organoid platform that consists of patient-specific tumor organoids is currently in process. It is intended to offer the opportunity to test the efficiency of drug therapies designed based on genetic profiling. Skardal et al. generated a circulatory system with multiple tissue organoid sites by using microfluidic chip devices and used them to visualize and track tumor progression and kinetics of metastasis formation to distant site in vitro [
132,
133]. In combination with the even more complex body-on-a-chip platform, these personalized on-a-chip systems will be improved even further [
132,
133].
This very promising technology offers great potential for in vivo tumor-like model systems to enable personalized drug screenings before treating patients and monitoring of organ systems in the OoC device for side effects at the same time. This new technology is still in development but has the potential to improve cancer treatment outcomes and patient care dramatically.
In summary, there are various suitable preclinical cancer models each with its own limitations. Despite this, these models are an attractive alternative or addition and have the potential to augment genetic and multi-omics approaches when considering ITH for precision cancer medicine.