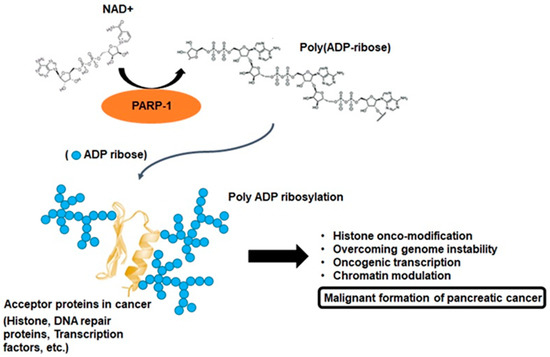
Oxidative phosphorylation is the basic metabolism responsible for the process of generating energy in the mitochondria, and as a result of this metabolism, essential compounds are also synthesized that are beneficial for the cell to survive [
18]. However, the result of this metabolism also causes the cells to undergo oxidative stress due to the production of reactive species following the activation of mitochondrial enzymes [
19]. Reactive oxygen species (ROS) account for a high proportion among these reactive species within the cells, and low concentration levels (<5%) of these species can be involved in benign metabolic events such as cell signaling, enzyme activation, and gene expression [
19,
20]. However, the loss of equilibrium between ROS and endogenous antioxidant species induces intracellular oxidative stress and alteration and damage to several intracellular molecules, including DNA, RNA, lipids, and proteins, even leading to the progression of apoptosis [
19]. Nevertheless, cancer cells can rather function positively under higher levels of oxidative stress conditions, which indicates the possibility that unique adaptive responses to oxidative stress would be an essential factor contributing to malignant transformation, including DNA damage, cell–cell adhesion, and signaling for sustained cancer cell survival [
20]. Regulating the balance or imbalance of intracellular ROS to maintain cellular functions is a well-established hallmark of pancreatic cancer cells. Maintaining a moderately higher level of ROS production than that in normal cells in the tumor microenvironment can act as a signaling molecule to promote the mutation of genomic DNA or enhance the proliferation of pancreatic cancer cells [
21]. On the one hand, excess ROS can cause irreversible oxidative damage, induce cell death through apoptosis, necrosis, and autophagy, and render pancreatic cancer cells susceptible to extracellular turbulences such as chemotherapy and radiation therapy [
20,
21]. Under such a critical environment, the functioning of the antioxidant program protects the pancreatic cancer cells from irreversible oxidative damage. The antioxidant program is driven by defense through enzymatic antioxidants, including the detoxification of secondary metabolites, as well as the direct removal of the electrophile itself [
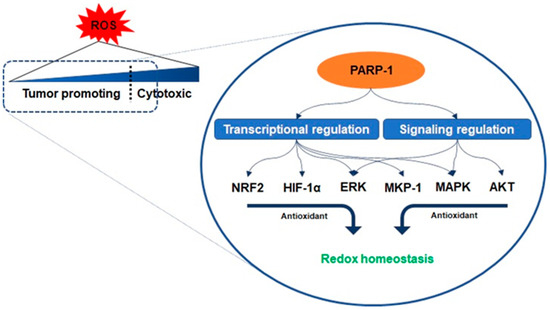
22]. Interestingly, PARP-1 controls a core role in the areas of such active utilization of oxidative stress conditions similar to a double-edged sword.
Antioxidant enzymes are under the transcriptional control of nuclear factor erythroid-related factor 2 (NRF2), a basic leucine zipper protein, and respond to physiological changes between intracellular redox actions for maintaining cellular homeostasis [
23]. Because PARP-1 promotes the interaction of antioxidant response elements with NRF2 and NRF2-partners, their interaction with PARP-1 enables the entire transcriptional activity of NRF2 [
24]. Another counteracting mechanism responsible for oxidant-involving PARP-1 is denoted by its interaction with hypoxia-inducing factor 1-alpha (HIF1-α), which undergoes activation during hypoxia-triggered redox imbalance [
25,
26]. PARP-1 forms a co-transcriptional activator with HIF1-α and enables the expression of the genes required for the maintenance of glutathione homeostasis, such as heme oxygenase-1 and glucose transporter 1, controlled in a PARP-1-dependent manner in a hypoxic response element promoter region [
25]. Furthermore, recent studies have reported that the signaling on ROS is dependent on PARP-1 activation related to the regulation of protein kinase B (AKT) [
27,
28,
29]. Cancer cells with KRAS mutation display constitutive activation of the AKT pathways, resulting in ROS production [
29]. As mentioned above, low concentration levels of ROS can be involved in benign metabolic events, but excessive ROS induces intracellular oxidative stress, such as damage to several intracellular molecules, leading to apoptosis [
19,
20]. The cascade in the AKT pathway, phosphatidylinositol 3, acts as a redox sensor and phosphorylates AKT to induce the active form, and AKT stimulates oxidative metabolism and forkhead box O-dependent catalase inhibition, contributing to the accumulation of hydrogen peroxide, however, PARP-1 activation and PAR synthesis inhibit the mTOR complex 1 signaling pathway and possibly modulate the mTOR complex 2, resulting in AKT downregulation [
27,
30,
31]. The increase in the levels of oxidants enhances the expression of the extracellular signal-regulated kinase (ERK) followed by the regulation of oxidant levels in the cells [
27]. PARP-1 involves the transcription of ERK and mitogen-activated protein kinase phosphatase 1 (MKP-1) in these signaling events, thereby regulating mitogen-activated protein kinase (MAPK) [
32]. Its regulation for the downregulation of MKP-1 blocks the dephosphorylation of the tyrosine and threonine residues of MAPK, which are activated upon acute oxidant exposure [
32]. In other words, PARP-1 is involved in blocking the dephosphorylation of the tyrosine and threonine residues in MAPK, thus contributing to the induction of sustained survival signals even under the increase in ROS () [
25,
32].