Hepatocellular cancer (HCC), the most common primary liver tumor, has been gradually growing in incidence globally. The whole-genome and whole-exome sequencing of HCC has led to an improved understanding of the molecular drivers of this tumor type. Activation of the Wnt signaling pathway, mostly due to stabilizing missense mutations in its downstream effector β-catenin (encoded by CTNNB1) or loss-of-function mutations in AXIN1 (the gene which encodes for Axin-1, an essential protein for β-catenin degradation), are seen in a major subset of HCC.
1. The Wnt–β-Catenin Signaling Pathway
The protein later termed Wnt1 was first identified almost 40 years ago in the context of its proto-oncogenic nature [1,2]. Subsequent studies have characterized Wnt1 itself, as well as other highly conserved components of Wnt signaling, as a key mediator involved not only in tumorigenesis, but also in the fundamental cellular processes governing embryonic development and adult tissue homeostasis [3,4]. Yet, the vital role of aberrant Wnt signaling in cancer initiation and progression remains one of the most intriguing and vital themes in the field. The Wnt pathway involves a multitude of components, including ligands, receptors, and co-receptors acting in autocrine, paracrine, and endocrine fashion to regulate the processes of cell fate determination, proliferation, and polarity, among others [2,4,5]. Structural and functional classification has indicated the existence of several distinct Wnt signaling pathways, which can be broadly subdivided based on the involvement of β-catenin. β-Catenin-dependent canonical Wnt signaling remains arguably the most investigated branch.
In the canonical pathway, the control of the Wnt-dependent cellular processes is achieved by a tight regulation of the amount of β-catenin—a transcriptional co-activator and a regulator of cell–cell adhesion. Normally, in the absence of Wnt signals, cytosolic levels of β-catenin remain low due to continuous proteasomal degradation of the protein, initiated by its destruction complex. The complex, composed of the scaffold Axin, tumor-suppressor adenomatous polyposis coli (APC) gene product, and diversin, also includes two kinases, casein kinase 1 (CK1), and glycogen synthase kinase 3 (GSK3), which sequentially phosphorylate β-catenin, priming it for recognition by the ubiquitin ligase β-TrCP [1]. In the absence of negative regulation, the glycosylation and palmitoylation of Wnt glycoproteins allows their biological activity to in turn activate the Wnt–β-catenin signaling. The cascade is induced by the binding of secreted Wnts to the seven transmembrane G-protein-coupled Frizzled (Fz) receptors located at the plasma membrane [5]. The binding initiates the formation of a multicomponent complex consisting of Wnt ligand, Frizzled, and its co-receptor LRP (low-density lipoprotein receptor-related protein) 6 or 5 [6]. This, in turn, signals for the recruitment of Dishevelled (Dvl), and results in the phosphorylation of LRP5/6, thereby providing a docking site for the Axin and tethering it to the cell membrane, which eventually renders the β-catenin destruction complex inactive. Thus, the presence of Wnt ligands interferes with the sequestration of β-catenin and its subsequent ubiquitination, thereby stabilizing the protein in cytoplasm. This allows for the nuclear translocation of β-catenin, where it triggers the expression of Wnt-induced genes (i.e., Cyclin D1, c-Myc, vascular endothelial growth factor (VEGF), interleukin-8 (IL-8), etc.) by acting as transcriptional co-activator in conjunction with T-cell factor (TCF) and lymphoid enhancer factor (LEF) DNA-binding proteins [7].
2. Wnt–β-Catenin Signaling in Liver Pathophysiology
The central role of the canonical Wnt–β-catenin signaling pathway in multiple aspects of normal cell functioning and in pathobiological processes is especially eminent in liver [8,9,10,11]. There, β-catenin orchestrates embryonic development, patterning, adult tissue metabolism, proliferation, and regeneration. While discussing the many facets of β-catenin signaling as a component of the Wnt pathway is outside the scope of the current review, we would like to remind the readers of a few pertinent concepts that are also relevant in hepatocellular cancer (HCC).
2.1. Wnt–β-Catenin Signaling in Hepatic Development
β-Catenin was first reported to be active in normal mouse and chick embryonic liver development almost two decades ago [
12,
13,
14]. β-Catenin was seen to be active in stages of hepatic development which showed proliferating hepatoblasts and immature hepatocytes. When mouse embryonic liver cultures were propagated in the presence of antisense oligonucleotides against the β-catenin gene, there was a notable deficit in the resident cell proliferation. This was later verified by conditional deletion of the β-catenin gene or via activation of β-catenin through APC gene loss from mouse hepatoblasts in vivo [
15,
16]. In addition to these observations, both in vitro and in vivo studies showed a dramatic compromise in hepatocyte maturation. This was seen as the maintenance of hepatoblast markers in the hepatocytes in the β-catenin absent or knocked-down livers, as well as by deficient markers of mature fetal hepatocytes, including glycogen [
16]. Thus, β-catenin plays a role in both the proliferation of immature hepatocytes and hepatoblasts during earlier stages of hepatic development, but plays an equally important role in the maturation of immature hepatocytes during later stages. These temporal targets of β-catenin include c-myc and cyclin-D1 for proliferation, as well as CEBPα and as-yet unknown targets, which are likely distinct from its well-known zone-3 targets in adult liver [
16]. It is also worth mentioning that, after birth, there is a postnatal growth spurt in livers from postnatal day 5 to about 25 days, after which the liver is mostly quiescent, showing minimal hepatocyte turnover [
17]. β-Catenin signaling is also a major contributor of the postnatal wave of hepatocyte proliferation, and in its absence there is a decreased growth spurt which leaves liver-specific β-catenin knockout mice with around 15% lower liver-weight to body-weight ratio (LW/BW).
2.2. Wnt–β-Catenin Signaling in Liver Regeneration
Livers possess a unique feature of regeneration following surgical resection or toxicant-induced injury to regain its lost mass within days to weeks. The liver does so without any progenitor cell activation but via the replication of resident hepatocytes (and other cells) in the liver [
18]. Wnt–β-Catenin signaling has been shown to be a key component of the normal molecular machinery of the liver following surgical resection [
19]. Within hours of two-thirds hepatectomy, there is a nuclear translocation of β-catenin in hepatocytes and the appearance of β-catenin–TCF complex [
20,
21]. This is sustained for almost the first 48 h of regeneration. Using several genetic knockout mouse models, it appears that Wnt2 and Wnt9b are massively upregulated in hepatic sinusoidal endothelial cells and less so in monocytes/macrophages at 12 h after hepatectomy (earliest time point examined through individual cell-type isolation after surgery), followed by the engagement of Fzd-LRP5/6, resulting in the activation of β-catenin–TCF4 to regulate cyclin-D1 gene transcription [
19]. The increased cyclin-D1 observed during this time allows for hepatocyte G1–S phase transition and eventually contributes to timely hepatocyte proliferation and the recovery of hepatic mass [
22]. The absence of Wntless from endothelial cells (and less so macrophages) or the absence of LRP5 and 6 from hepatocytes or the absence of β-catenin from hepatocytes, all lead to a notable deficit in cyclin-D1 expression and a dramatically lower hepatocyte proliferation at 40–48 h after two-thirds hepatectomy [
23,
24,
25,
26,
27]. Livers eventually recover in all models, despite a notable delay in restitution, and the mechanisms allowing for recovery in the absence of Wnt–β-catenin signaling remain unknown at this time. A similar role of the pathway during hepatocyte proliferation has also been reported after injury from acetaminophen, carbon-tetrachloride, diethoxycarbonyl dihydrocollidine, choline-deficient ethionine supplemented diet, and in Mdr2 knockout mice, making Wnt–β-catenin signaling a global hepatic repair pathway [
28,
29,
30,
31,
32,
33].
Intriguingly, a recent study also showed an important role of the Wnt–β-catenin pathway in serving a dual role of not only inducing hepatocyte proliferation but also maintaining hepatocyte function during liver regeneration after surgical resection, as well as after acetaminophen-induced injury and repair. Using single-cell RNA-sequencing, Walesky et al. showed a clever “division of labor” by the hepatocytes in the remnant liver following surgery or toxicant injury [
34]. This strategy allows liver to maintain function even while it is proliferating, as distinct subsets of hepatocytes acquire proliferative versus hepatocyte-function phenotype, as shown by gene expression studies. Intriguingly, both these functions are regulated by the Wnt–β-catenin pathway; the cell source of the Wnt for regulating the hepatocyte function by β-catenin appears to be macrophages and not sinusoidal endothelial cells, which are likely the source of Wnts for β-catenin activation in hepatocytes for proliferative function.
2.3. Wnt–β-Catenin Signaling in Liver Zonation
Another unique characteristic of the liver is the expression of unique genes by the hepatocytes based on their location within a microscopic hepatic lobule. This disparate gene expression allows for the hepatocytes to perform distinct functions that are necessary for the delivery of optimal hepatic output in terms of metabolism, synthesis, and detoxification, which are the broad categories of around 500 functions that hepatocytes perform to maintain health and homeostasis. Toward this end, Wnt–β-catenin signaling is known to be the major regulator of the expression of genes in the zone-3 or pericentral region of the metabolic lobule [26,35,36]. These genes belong to the category of glutamine synthesis, glycolysis, lipogenesis, ketogenesis, bile acid synthesis, heme metabolism, and xenobiotic metabolism. Some of these target genes include Glul, which encodes glutamine synthetase (GS), and is specifically localized to 1–2 layers of hepatocytes around the central vein [37]. To prevent ammonia from leaving the liver, the zone-3 hepatocytes are efficient in its uptake and the high levels of GS in these cells are responsible for condensing ammonia to glutamate, leading to the formation of glutamine. Thus, intracellular levels of glutamine are highest in zone-3 hepatocytes. Some of the other key targets of β-catenin in zone-3 hepatocytes include Axin-2, Lect2, Cyp2e1, Cyp1a2, and others. Recently, choline transporter organic cation transporter 3 was also shown to be a target of the Wnt–β-catenin signaling, which led to the increased uptake of choline by HCC to promote phospholipid formation and DNA hypermethylation, and contributing to hepatocyte proliferation [38]. In fact, several of these β-catenin targets are upregulated in liver tumors where β-catenin signaling is highly activated in both preclinical models and in patients. Conversely, genetic knockout models that lack Wnt secretion from endothelial cells, lack LRP5 and 6 on hepatocytes, or lack β-catenin in hepatocytes, all lack zone-3 targets of the Wnt–β-catenin pathway [23,24,26,27,36]. Wnt2 and Wnt9b appear to be the major drivers of zonated β-catenin activation, and appear to be within the endothelial cells lining the central vein [39].
Thus, broadly, β-catenin seems to be playing a role in hepatocyte proliferation in physiological states including hepatic development (prenatal and postnatal) and liver regeneration (surgical and injury-driven), as well as in regulating hepatocyte functions including basally in the hepatocytes contained in zone-3 of the metabolic lobule. It is pertinent to mention the existence of regulators of the Wnt–β-catenin signaling that have been shown to play a role in the aforementioned hepatic processes. Factors like R-spondins and their receptors LGR4/5 have been shown to potentiate the effects of the Wnt–β-catenin pathways and have been specifically shown to positively impact the processes of both liver regeneration and metabolic zonation [40,41].
3. Targeting β-Catenin for HCC Treatment
3.1. Proof-of-Concept Studies
Because β-catenin is active in a notable subset of HCCs, and is also considered a trunk mutation, its inhibition could have a major impact on the treatment of a subset of these tumors. Several proof-of-concept studies in HCC, both in vitro and in vivo, have demonstrated the relevance of inhibiting β-catenin as a treatment strategy for HCC. siRNA-mediated
CTNNB1 knockdown resulted in a marked decrease in the viability and proliferation of human hepatoma cells in vitro [
79]. Similarly, suppressing β-catenin via gamma-guanidine-based peptide nucleic acid antisense also reduced the viability, proliferation, metabolism, and survival of cells of an HCC line [
80]. Interestingly, inhibition of β-catenin signaling also resulted in the diminished secretion of angiogenic factors, implying the dual positive effect of such suppression [
80]. The DsiRNAs-mediated knockdown of β-catenin mRNA led to a significant decrease of tumor burden in mice bearing ectopic tumors originating from either Hep3B or HepG2 cells [
81]. Using a chemical carcinogen (diethylnitrosamine) and tumor promotion (phenobarbital) model which selectively leads to
Ctnnb1-mutation-driven HCC, β-catenin inhibition using locked nucleic acid antisense (LNA) had a profound impact on tumor development [
82]. More recently, using Kras–β-catenin-driven HCC (which highly resembles the Met–β-catenin model), β-catenin was inhibited using EnCore lipid nanoparticles loaded with a Dicer substrate small interfering RNA targeting
CTNNB1. This led to a notable decrease in tumor burden, also demonstrating β-catenin to be a highly relevant target in HCC for cases driven by
CTNNB1 mutations.
3.2. Where to Target Wnt–β-Catenin Signaling in HCC
The most important mechanism of β-catenin activation in HCC are the mutations in CTNNB1 and the mutations in AXIN1. While there have been several other mechanisms identified to modulate β-catenin signaling, including the upregulation of certain Wnt genes, Frizzled genes, and epigenetic loss of negative regulators like DKK and FRPs and others, their true relevance remains unclear since Wnt–β-catenin signaling, like other signaling pathways, is able to regulate its overall activity via robust post-translational mechanisms. However, mutations in CTNNB1 or AXIN1 deem the β-catenin protein non-degradable and hence cannot be regulated by the normal mechanisms, which converge on β-catenin degradation to control the signaling pathway activity. This also suggests that several classes of Wnt inhibitors will not work in HCCs because they inhibit or impair Wnt activity upstream of the observed mutations in CTNNB1 or AXIN1. Hence, the suppression of β-catenin itself using the RNA-based therapies discussed in the preceding section, or those impairing β-catenin nuclear translocation, impairing its interaction with TCF4 or preventing the β-catenin–TCF complex from transactivating target genes, would be most effective in treatment of some subsets of HCC. Finally, identifying unique opportunities related to β-catenin signaling in HCC is important, as it may help in selecting or excluding the right group of patients, and may help to identify innovative opportunities to target other mechanisms that are intimately related to β-catenin activation unique to HCC.
3.3. How to Target β-Catenin in HCC
Targeting β-catenin itself using RNA-based therapies is highly desirable. Several classes of siRNA- and antisense-based therapies have been described for use against β-catenin. The use of EnCore lipid nanoparticles along with Dicer substrate small interfering RNAs is especially innovative because it can be modified to specifically deliver the payload to liver tumors, and the safety of their use has been shown in patients [
83]. Others such as peptide nucleic acid antisense, locked nucleic acid antisense, and other modalities have been reported, and may have eventual clinical use [
80,
81,
82].
There may be an opportunity to identify the mechanisms of the nuclear transport or nuclear export of β-catenin. Targeting molecules that cargo β-catenin to the nucleus or activate its export out of the nucleus could have efficacy in the treatment of β-catenin-mutated HCCs. Pegylated interferon-α2a (peg-IFN), previously a first-line therapy for hepatitis C virus (HCV) patients, was shown to induce the levels of Ran-binding protein 3 (RanBP3), which is known to export β-catenin out of the nucleus [
84]. Peg-IFN treatment was also shown to induce association between RanBP3 and β-catenin, and led to decreased TopFlash reporter activity that was abrogated by siRNA-mediated RanBP3 knockdown. In vivo, peg-IFN treatment led to increased nuclear RanBP3, decreased nuclear β-catenin and cyclin D1, and decreased GS, and eventually led to decreased tumor cell proliferation.
The use of small-molecule inhibitors that interfere with its interactions with TCF or other relevant co-factors or components of the transcriptional complex would be highly desirable. However, a high specificity of the small-molecule inhibitors will be required because of the overlap of the β-catenin–TCF4 binding site, and with the binding sites for APC and E-cadherin [
85]. Even though a number of the identified compounds showed selectivity of inhibition in vitro (e.g., PKF115-584, CGP049090, and PKF118-310), none of them has entered clinical trials [
85]. PR1-724, the next-generation compound of the original small-molecule ICG-001, interferes with β-catenin–TCF4 interactions with CBP, a histone acetyltransferase essential for transcriptional function of the complex [
86,
87]. PRI-724 has been shown to be safe in patients with HCV-related cirrhosis, and may be of high relevance in the treatment of subsets of HCC with known mutations in
CTNNB1 [
88].
3.4. Unique and Exploitable Aspects of Targeting β-Catenin in Subsets of HCC
In addition to a general role of β-catenin in regulating tumor cell proliferation, survival, and angiogenesis, there are specific and unique aspects of β-catenin activation due to mutations in HCC which can have notable biological and therapeutic implications—especially related to a step towards precision medicine.
3.4.1. Role of β-Catenin in Tumor Immune Evasion
ICIs have revolutionized the treatment of many tumors, including HCC as can be seen by the FDA approval of nivolumab and pembrolizumab as second-line therapy and of atezolizumab (anti PD-L1) plus bevacizumab (anti-VEGFA), as first line treatment for unresectable HCC [
78]. However, there are no available biomarkers which predict either the efficacy or lack thereof to ICIs. Clinical response to ICIs, most of which are T-cell-based therapies, depend on the presence of a CD8
+ T cell inflamed environment and chemokines and interferon signature within the tumor [
89]. Intriguingly, activation of β-catenin signaling has been linked to immune evasion in tumors such as melanoma through T-cell exclusion from tumors [
90]. This is shown in our own analysis as well (). Several mechanisms underlie this observation, including the effect of β-catenin activation on CD8
+ T cell priming and infiltration by acting on Batf3-lineage CD103
+ dendritic cells (DCs) and decreasing CCL4 production by inducing the expression of transcription repressor ATF3 [
91]; disruption of Foxp3 transcriptional activity, key for development and function of regulatory T cells [
92]; and increased Treg survival, which can reduce CD8
+ T cell proliferation [
93]. HCCs with β-catenin activation have been linked to immune cell exclusion [
94,
95]. We have shown that
CTNNB1-mutated HCCs are resistant to anti-PD-1 [
68], and hence may benefit from the inhibition of β-catenin or its downstream effectors to sensitize these tumors to ICIs.
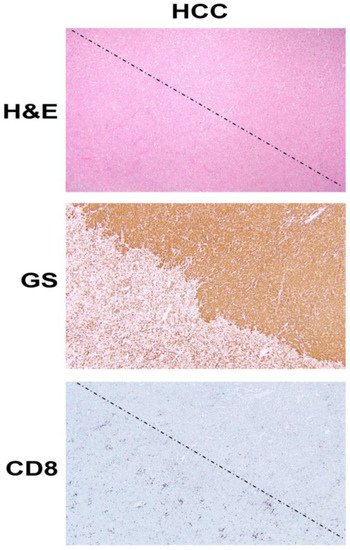
Figure 1. β-Catenin activation in HCC reduces CD8 T cell infiltration in the tumor. The top panel shows histology of explanted liver for hepatocellular cancer (HCC) showing the presence of two distinct tumors (separated by a dotted line) which are otherwise difficult to distinguish and demarcate by hematoxylin and eosin (H&E) staining (100×). The middle panel shows the immunohistochemistry of the adjacent tissue section to the top panel, for glutamine synthetase (GS), a surrogate marker of β-catenin activation due to mutations in CTNNB1. The staining for GS shows the presence of uniform positive staining in the upper-right part which decorates a β-catenin-active HCC, whereas the lower-left tumor is negative for this stain. Immunohistochemistry for CD8 for a subset of T cells, in the section adjacent to those shown in the top and middle panels, shows a general dearth of positive cells in the top-right (β-catenin-active) tumor, while there are notably more CD8-positive cells in the lower left or in the non-β-catenin-active HCC. The two tumors are separated by a dotted line.
One additional relevant mechanism in the liver might be through a known interaction of β-catenin with NF-κB in the hepatocytes and liver tumor cells [
96]. This inhibitory association between the p65 subunit of NF-κB and β-catenin prevents NF-κB activation even when appropriate upstream effectors of NF-κB are present. In this study, we also showed that this association led to reduced p65-luciferase reporter activity when constitutively active β-catenin was transfected in hepatoma cells. Furthermore, β-catenin mutated HCCs showed decreased p65 nuclear translocation. Knowing that NF-κB signaling plays a major role in inducing inflammatory milieu [
97], its suppression brought about by stable β-catenin due to mutations in
CTNNB1 may be one additional contributor of an immune-deficient tumor microenvironment which may in turn lead to resistance to ICIs.
3.4.2. Role of β-Catenin in Regulating Tumor Metabolism Through mTORC1 in HCC
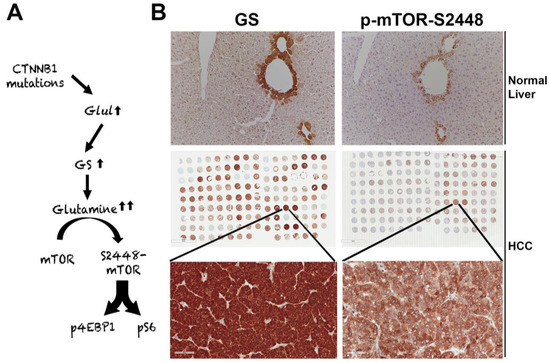
The suppression of β-catenin in CTNNB1-mutant liver tumors decreases tumor burden in many models [71,82]. We made a unique discovery of how this response was mediated by the regulation of mTORC1 by β-catenin [98]. The Wnt–β-catenin pathway transcriptionally regulates the expression of Glul, which encodes GS in hepatocytes in zone-3 of the hepatic lobule [37], and leads to the highest glutamine in zone-3 hepatocytes [99] (A,B). Glutamine directly phosphorylates mTOR at serine-2448 in lysosomes [100]. We identified p-mTOR-S2448 (active mTORC1) [101] in zone-3 hepatocytes basally, which was absent in hepatocyte-specific knockout (KO) of β-catenin, Wnt co-receptors LRP5-6, and GS (B). We also found by immunohistochemistry (IHC) that HCCs with CTNNB1 mutations are simultaneously positive for GS and p-mTOR-S2448 in preclinical models and patients (B). We also showed a dependence of the CTNNB1-mutated HCCs to mTORC1 by their susceptibility to mTOR inhibition by rapamycin in a preclinical model. This may be a novel way to target β-catenin mutated liver tumors in patients until anti-β-catenin therapies become a reality.
Figure 2. Unique mTORC1 addiction of CTNNB1-mutated HCCs due to glutamine. (A) The unique axis of mTORC1 activation in β-catenin gene mutated HCCs due to overexpression of GLUL, the gene encoding for glutamine synthetase (GS), which generates glutamine from ammonia and glutamate, and in turn glutamine activates mTORC1 in lysosomes. (B) The top panel shows immunohistochemistry for GS and p-mTOR-S2448 in adjacent sections from a normal mouse liver. Both proteins are localizing exclusively to zone-3 hepatocytes in the immediate proximity to the central vein (200×). The whole slide scans (middle row) of two adjacent tissue microarrays of human HCC samples stained for the same antibodies against GS and p-mTOR-S2448 also shows several HCCs to be simultaneously positive for GS and p-mTOR-S2448. A representative tissue array sample is magnified (400×) to show GS and p-mTOR-S2448-positive HCC (bottom panels).
This entry is adapted from the peer-reviewed paper 10.3390/cancers13081830