Atherosclerosis is a chronic inflammatory process within the arterial wall.
- prediabetes
- atherosclerosis
- metabolic syndrome
- obesity
- endothelial dysfunction
- exosome
- microRNA
1. Introduction
Contrary to the common belief that atherosclerosis is a disease of the elderly, the disease can begin surprisingly early, even before the age of 14 [1]. The modern age of gourmet foods, overeating, and underexercising has caused many people to develop metabolic syndromes that eventually lead to prediabetes and diabetes. Genetic factors probably play a role in this process. The likelihood of developing diabetes increases in subjects with a family history of diabetes [2]. Diabetes is one of the critical factors in the development of atherosclerosis. This fact is even relevant to the pandemic of coronavirus disease 2019 (COVID-19), as recent reports showed that patients with diabetes mellitus (DM) and atherosclerosis are at a greater risk for severe COVID-19 complications [3]. Therefore, it is essential to understand the mechanism underlying the development of atherosclerosis as diabetes progresses.
Numerous studies indicated that prediabetes can cause cardiovascular disease (CVD) [4,5]. Moreover, the burden of coronary atherosclerosis in prediabetic patients is more significant than that in normal people. Notably, the burden of atherosclerosis appeared even before the clinical manifestations of DM as seen in [4]. For example, lipid-rich plaques that congest blood vessels were already found in coronary lesions in the prediabetic state [4,6]. In addition, inflammation and vasoconstriction, which promote atherosclerosis in the coronary arteries, were observed in the prediabetic state [7,8].
2. The Effect of the Release of Inflammatory Factors Caused by Obesity on CVD
Atherosclerosis is a chronic inflammatory process within the arterial wall [9]. Atherosclerosis is promoted when cholesterol-rich atherosclerotic Apo-B lipoproteins, such as low-density lipoproteins (LDL), very-low-density lipoproteins, and intermediate-density lipoproteins, enter into the subendothelial space via the endothelial passage (transcytosis) and accumulate in plasma [9]. The transcytosed Apo-B lipoproteins infiltrate macrophages and T cells to interact with the arterial wall cells, activating the inflammatory response [9,10]. Inflammation caused by obesity may accelerate atherosclerosis [11]. Adipose tissue is crucial in CVD caused by obesity [12]. In this situation, both fat cells and activated macrophages from the adipose tissue can produce various cytokines [12] (see Figure 1). These cytokines include adipokines induced by inflammation, such as leptin, adiponectin, tumor necrosis factor-alpha (TNF-α), interleukin 1 (IL-1), and IL-6; coagulants, such as PAI-1; vasoactive substances, such as leptin, angiotensinogen, and endothelin; and molecules such as FFA, TNF-α, and resistin that may cause insulin resistance. Table 1 shows the blood levels of several inflammatory factors associated with the development of atherosclerosis in prediabetes and diabetes.
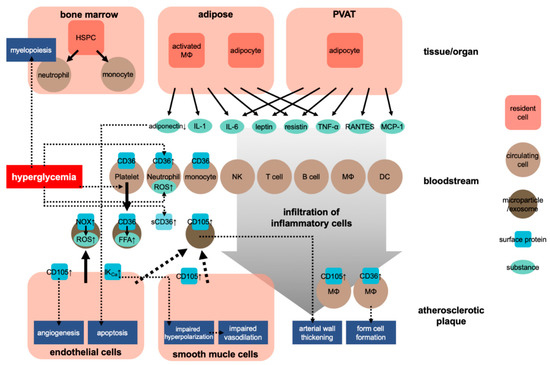
Figure 1. Development of atherosclerosis in obesity. HSPC, hematopoietic stem and progenitor cells; NK, natural killer cell; MΦ, macrophage; DC, dendritic cell; sCD36, soluble CD36; ↑, upregulation.
Table 1. Comparison of the factors contribute to atherogenesis between prediabetes and diabetes.
| Normal | Prediabetes | Diabetes | Refs | |
|---|---|---|---|---|
| Fasting glucose (mg/dL) | 86.4 ± 8.6 | 93.4 ± 11 | 120.2 ± 19 | [26] |
| HbA1c (%) | 5.3 ± 0.2 | 6.0 ± 0.3 | 7.1 ± 0.5 | [26] |
| Total cholesterol (mg/dL) | 189–202 | 196–203 | 189–200 | [26,27] |
| HDL cholesterol (mg/dL) | 48–55 | 46–48 | 39–45 | [26,27] |
| LDL cholesterol (mg/dL) | 120–121 | 126–129 | 121–126 | [26,27] |
| Triglycerides (mg/dL) | 86–111 | 93–126 | 120–147 | [26,27] |
| hs-CRP (mg/L) | 1.4–2.1 | 2.4–3.4 | 4.0–4.5 | [26,27] |
| esRAGE (ng/mL) | 0.52 ± 0.26 | 0.32 ± 0.18 | 0.3 ± 0.19 | [26] |
| S100A12 (ng/mL) | 5.35 ± 3.38 | 7.13 ± 5.4 | 8.41 ± 4.44 | [26] |
| Adiponectin (μg/mL) | 9.52 ± 0.49 | 6.15 ± 0.49 | 6.57 ± 0.457 | [28] |
| IL-6 (pg/mL) | 1.77 ± 0.23 | 2.00 ± 0.14 | 2.84 ± 0.62 | [29] |
| Resistin (ng/mL) | 5.11 ± 1.56 | 9.16 ± 3.06 | 14.5 ± 5.31 | [30] |
| TNF-α (pg/mL) | 1.31 (0.69–2.25) |
1.68 (0.79–2.01) |
1.41 (0.83–1.86) |
[31] |
| White blood cell (103/μL) | 6.4 ± 1.6 | 7.1 ± 1.8 | 7.2 ± 1.8 | [26] |
| Intima-media thickness (mm) | 0.67 (0.6–0.73) |
0.75 (0.65–0.78) |
0.78 (0.7–0.92) |
[26] |
| Pulse wave velocity (m/sec) | 7.1 ± 1.7 | 7.6 ± 1.6 | 8.6 ± 1.7 | [26] |
| Coronary plaque progression (odds ratio) | - | 1.338 | 1.635 | [32] |
| Coronary artery calcification (odds ratio) | - | 1.253 | 2.215 | [33] |
| Atherosclerotic cardiovascular disease events (%) | 14.24 | 17.81 | 30.40 | [27] |
HbA1c, glycated hemoglobin A1c; HDL, high-density lipoprotein; LDL, low-density lipoprotein; hs-CRP, high-sensitivity C-reactive protein; esRAGE, endogenous secretory advanced glycation end-products; IL-6, interleukin-6; TNF-α, tumor necrosis factor-α.
IL-1 signaling involves the type I IL-1 receptor (IL-1R/IL-1R1) that can heterodimerize with the IL-1R accessory protein (IL-1RAcP) [13,14]. IL-1 receptor antagonist (IL-1Ra) is an anti-inflammatory cytokine that can compete with the proinflammatory cytokine IL-1 to bind to IL-1R. The relative occupation rate of the IL-1R1–IL-1RAcP receptor complex for IL-1 agonist or IL-1Ra controls the activation of the inflammatory signal [15,16]. In the obese condition, these cytokines released into the adipose tissue’s circulatory system can stimulate the production of C-reactive protein in the liver [17]. The level of the thrombosis molecule PAI-1 also increases before the development of obesity, whereas the level of adiponectin, which is only produced by fat cells, decreases [18]. Leptin is a crucial regulator of food intake and is a critical vasoactive substance produced by fat cells [19]. Other molecules produced by fat cells, including prostaglandin, adiponectin, and resistin, can influence metabolic function and lead to cardiovascular end-organ damage [20].
Regarding the effect of obesity on the cardiovascular system, researchers have also focused on the role of perivascular adipose tissue (PVAT). PVAT is an active endocrine organ that anatomically wraps the blood vessels and plays an important role in the pathogenesis of CVD. PVAT can secrete various adipokines, cytokines, and growth factors that inhibit or stimulate CVD development [21,22] (Figure 1). Dysfunction of PVAT can cause inflammation and oxidative stress and can decrease the production of vasoprotective adipocyte-derived relaxation factors and paracrine factors, such as resistin and leptin, and that of cytokines, such as IL-6 and TNF-α, and chemokines, such as RANTES and MCP-1 (also known as CCL5 and CCL2, respectively) [21]. These adipocyte-derived factors can trigger and coordinate the infiltration of inflammatory cells, such as T cells, B cells, and NK cells. Protective factors such as adiponectin can reduce the generation of NADPH oxidase superoxide and promote the bioavailability of nitric oxide (NO) in the blood vessel wall. Inflammatory molecules (such as IFN-γ or IL-17) can cause fibrosis in the endothelium, vascular smooth muscle cells, and adventitia, leading to the induction of vascular oxidase and eNOS dysfunction in cells [23,24]. These conditions lead to the occurrence of vascular dysfunction caused by dysfunctional perivascular fat. These mechanisms play a critical role in several CVDs, including atherosclerosis, hypertension, diabetes, and obesity [21,25].
3. Prediabetes Affecting Monocyte and Macrophage Activities That Lead to Atherosclerosis
For the progression of atherosclerosis, it is critical for the recruited monocytes to engulf LDL accumulated under the surface layer of blood vessels [34]. Hyperlipidemia in DM is known to increase the number of circulating neutrophils and monocytes through myelopoiesis [35].
Interestingly, Flynn et al. reported that even in the absence of DM or insulin resistance, the change in glucose level activated the bone marrow, leading to mononucleosis and accelerating atherosclerosis [36]. Meanwhile, transient intermittent hyperglycemia (TIH) caused hematopoietic stem and progenitor cells (HSPC) to proliferate and differentiate in the bone marrow, leading to a rapid expansion of common myeloid and granulocyte-macrophage progenitors and a subsequent white blood cell proliferation. In particular, TIH caused the proliferation of Ly6Chi mononuclear cells, which induce atherosclerosis more strongly than other monocytes. According to Graubardt et al., removing the circulating Ly6Chi monocytes reduces neutrophils that produce reactive oxygen species (ROS) [37], which is one of the critical factors of both DM and atherosclerosis [38]. Other risk factors found in prediabetic obesity, such as inflammation, dyslipidemia, oxidative stress, and signal transduction from adipose tissue macrophages, may also promote the expansion and differentiation of bone marrow HSPCs and, subsequently, leukocytosis [39,40,41].
On the other hand, glucose uptake by neutrophils after TIH is vital for driving CVDs [42,43]. Glucose transporter 1 (GLUT-1) is involved in glucose uptake; myeloid-restricted GLUT-1 deletion reduces glucose uptake by neutrophils and prevents TIH-induced myelopoiesis and atherosclerosis [36,43]. Flynn et al. found that when the glucose level is too high, neutrophils quickly reach their maximum glycolysis rate, leading to ROS production and protein kinase C (PKC) activation, which triggers the release of S100 calcium-binding proteins A8 and A9 (S100A8/A9). The release of S100A8/A9 initiates bone marrow production. S100A8/A9 proteins account for 40% of the neutrophil cytoplasmic protein, which is released upon inflammation and stimulates leukocyte recruitment, and it induces cytokine secretion [44,45]. S100A8/A9 is also upstream of the receptor for advanced glycation end-products (RAGE) signaling. The RAGE pathway plays a crucial role in hyperglycemia-induced myelopoiesis and TIH-induced mononucleosis. Koulis et al. discovered that RAGE mediation plays a vital role after a brief increase in blood sugar in atherosclerosis [46].
Another calcium-binding protein, S100A12, alias extracellular newly identified RAGE binding protein, is found in high concentrations in the blood of prediabetes and diabetes [26] (Table 1). As it facilitates the expression of adhesion molecules in the endothelial cells and the resulting migration of monocytes and macrophages, it is regarded as a pro-atherosclerotic factor [47]. Furthermore, the blood level of endogenously secreted RAGE, which can contribute to the neutralization of circulating RAGE ligands, is decreased in prediabetes [26]. This further facilitates the development of atherosclerosis. Therefore, targeting the RAGE pathway may be essential in eliminating hyperglycemia-induced atherosclerosis.
4. The Involvement of miRNAs in the Pathogenesis of Prediabetes and Atherosclerosis
In the previous section, we discussed the identification of extracellular vesicles express surface markers, such as CD36 and CD105, in prediabetic patients. These vesicles are likely involved in the development of atherosclerosis. CD36 is actively involved in fatty acid transport in macrophages. In addition to containing functional surface proteins, extracellular vesicles transport miRNAs in the bloodstream [96]. While some miRNAs are transported in an encapsulated form in vesicles, most miRNAs maintain their stability by forming a complex with other proteins during the transport in the bloodstream. This section discusses the mechanisms of miRNAs’ possible involvement in the pathogenesis of prediabetes and atherosclerosis.
4.1. Macrophage miRNAs Reducing the Production of Myeloid Cells and Suppressing Inflammation in Atheroma
Specific kinds of miRNAs induce anti-inflammatory effects. For example, the anti-inflammatory effect of miR-146b has been reported in a sepsis disease model and pneumonia [97,98]. In addition, miR-146b was reported to reduce inflammation via TRAF-6, a signal transducer in the NF-κB pathway, in a hypercholesterolemic condition that leads to atherosclerosis [99]. The expression level of miR-146b has been associated with HbA1c, a marker for high blood sugar level maintained over weeks/months [100]. Consistent with this finding, miR-146a expression is higher in DM patients than in prediabetic patients [101]. On the other hand, miR-99a, an anti-inflammatory miRNA, inhibits the production of inflammatory mediators, such as TNF-α, IL-6, IL-1β, and MCP-1 [102]. Moreover, the IL4-driven anti-inflammatory effects of miR-378a on macrophages have been suggested [103].
Interestingly, Bouchareychas et al. reported that macrophage exosomes ameliorated atherosclerosis by regulating hematopoiesis in the bone marrow and inflammation in atheroma through miRNA [104]. Mainly, the exosomes generated from naive bone marrow-derived macrophages (BMDM) contained anti-inflammatory miR-99a, miR-146b, and miR-378a [104]. It was found that exposure of IL-4 to BMDMs further facilitated the production of exosomes containing the anti-inflammatory microRNAs. These exosomal microRNAs inhibited the inflammation in atheroma by aiming at NF-κB and TNF-α signaling. Furthermore, the infusion of exosomes from IL-4 activated BMDM into mice can reduce hematopoiesis in bone marrow, thereby reducing both the number of macrophages in aortic root lesions and the number of atherosclerotic necrotic lesions.
The discovery that atherosclerosis development can be suppressed by exosomes containing miRNAs is quite sensational. Additionally, the high expression level of miR-146b in DM patients suggests that an anti-inflammatory response had already occurred in these patients. Thus, the administration of exosomes containing miR-99a and miR-378a to suppress the progression of atherosclerosis or inflammation is an anticipated clinical strategy.
4.2. Prediabetes Altering Expression of miRNAs in the Heart
Éva Sághy et al. found that prediabetes alters the expression of specific miRNAs that subsequently modulate their corresponding target mRNAs’ expression in the left ventricles of rats [105]. This study also found that miR-141 and miR-200c were upregulated, and that miR-200a, miR-208b, and miR-293 were downregulated in prediabetic patients. The authors predicted their target mRNAs computationally from the sequences of these miRNAs. They then verified the down-regulation of three mRNAs, i.e., the juxtaposed with another zinc finger protein 1 (JAZF1), Rap2c of the RAS oncogene family, and zinc fingers with KRAB and SCAN domain 1 (ZKSCAN1). The miRNAs were isolated from homogenized ventricular tissue in this study. Therefore, it is necessary to investigate the origin of the detected miRNAs. It would also be interesting to see whether exosomes containing these miRNAs are released into the bloodstream. While the miRNAs may be released from noncardiomyocytes, such as cardiac fibroblasts or vascular endothelial cells, some studies suggested that cardiomyocytes secrete the exosomes, and that the effect of the exosomes is derived from miRNAs [106].
As mentioned above, JAZF1 is one of the genes downregulated by miRNA in prediabetic patients. JAZF1 is a transcriptional cofactor involved in gluconeogenesis, lipid metabolism, and insulin resistance; it is related to JAZF1 expression in type 2 DM [107,108]. According to Li et al., JAZF1 prevents atherosclerosis by reducing the number of atherosclerotic plaques in the aortic sinus [107]. Therefore, the decreased expression of JAZF1 in prediabetes may be associated with the development of atherosclerosis.
4.3. miR-483 Causing Lipotoxicity, Insulin Resistance, and Impaired Endothelial Integrity
What we eat in the early stages of development affects the subsequent development of diabetes and atherosclerosis. Even more remarkably, the expression of a specific miRNA provides this effect. miR-483 can be partially mediated by the translational growth inhibition/differentiation factor 3. It can also regulate the adipocytes’ ability to differentiate and store lipids [109]. Partially mediated by translational growth inhibition/differentiation factor 3, a target of miR-483, miR-483 regulates the adipocytes’ ability to differentiate and store lipids. In summary, early life nutrition induces an increase in miR-483 expression, which in turn limits lipid storage in adipose tissue, causing lipotoxicity and insulin resistance and increasing the risk of metabolic diseases.
In addition, miR-483’s expression level is higher in M2-type macrophages and in the aortic wall of patients with type 2 DM [110]. Moreover, overexpressing miRNA-483 causes endothelial apoptosis and impairs endothelial regeneration [110]. Furthermore, serum miR-483 level is associated with DM and CVD [111]. It would be interesting to identify the tissue that causes the elevation of serum miR-483 and to examine whether the events occurring in endothelial cells and adipose tissue are associated with serum miR-483. In summary, excessive nutrition during early development may cause the upregulation of miR-483, leading to impaired endothelial integrity and subsequent CVDs. These possible effects of miR-483 upregulation should be examined in future studies.
(References would be added automatically after the entry is online)
This entry is adapted from the peer-reviewed paper 10.3390/ijms22084108