Anti-angiogenics currently used in cancer therapy target angiogenesis by two major mechanisms: (i) neutralizing angiogenic factors or their receptors by using macromolecule anti-angiogenic drugs (e.g., therapeutic antibodies), and (ii) blocking intracellularly the activity of receptor tyrosine kinases with small molecule (Mr < 1 kDa) inhibitors. Anti-angiogenics halt the growth and spread of cancer, and significantly prolong the disease-free survival of the patients. However, resistance to treatment, insufficient efficacy, and toxicity limit the success of this antivascular therapy. Published evidence suggests that four albumin-binding proteins (ABPs) (gp18, gp30, gp60/albondin, and secreted protein acidic and cysteine-rich (SPARC)) could be responsible for the accumulation of small molecule receptor tyrosine kinase inhibitors (RTKIs) in normal organs and tissues and therefore responsible for the side effects and toxicity associated with this type of cancer therapy.
- albumin-binding proteins
- albumin-drug complexes
- angiogenesis
- anti-angiogenic therapy
- endocytosis
- transendothelial transport
- therapeutic agents
1. Introduction
In animal models for human cancer, angiogenesis is a prerequisite for tumor growth beyond 2 mm3 [1]. The endothelial cell (EC) proliferation is stimulated by various tumor secreted angiogenic factors including vascular endothelial growth factor (VEGF) [2], platelet-derived growth factor (PDGF) [3], fibroblast growth factor (FGF) [4][5], and angiopoietins [6]. Angiogenic factors act via paracrine signaling when they are released by tumor and stromal cells or when they are mobilized from the extracellular matrix (ECM) [5]. The information conveyed by the angiogenic factors is transmitted to transmembrane tyrosine kinase receptors that are expressed on the abluminal surface of ECs lining the pre-existent blood vessel neighborhood of a tumor implant. The activation of these ECs causes degradation of the endothelial basal membrane and of the ECM, which facilitates the EC migration and proliferation, and a tube formation resulting in new vascular sprouts [7]. As the sprouting of new blood vessels from the pre-existing ones is a sine qua non condition of tumor progression and metastasis, inhibiting this process by using anti-angiogenesis therapeutic agents may halt the growth and spread of cancer [8][9].
Anti-angiogenic therapy is based on the concept that tumor vessels can be selectively targeted without affecting the normal organs’ vasculature [10], which is characterized by an extensive coverage with pericytes that can control the quiescent endothelial phenotype [11]. The anti-angiogenic drugs currently used in cancer therapy target the proliferating tumor ECs by two major mechanisms: neutralizing angiogenic factors or their receptors by using macromolecule anti-angiogenic drugs (e.g., therapeutic antibodies) or blocking the receptor tyrosine kinases intracellularly with small molecule (Mr < 1 kDa) receptor tyrosine kinase inhibitors (RTKIs) bound to albumin [12]. While some anti-angiogenic drugs inhibit the pathways that affect the initiation of tumor angiogenesis (e.g., the VEGF pathway), others impair the maintenance of the angiogenic process (e.g., the FGF pathway).
The encouraging study data on angiogenic factor-targeted therapies and their mechanisms of action in preclinical models have led to the translation of these therapies to the clinic (e.g., VEGF-targeted therapies) [13][14][15]. However, anti-angiogenic therapies have shown limited efficacy in the clinical management of various types of cancer. One reason for this seems to be the difference between the highly proliferative experimental tumors supported by a new immature highly angiogenic microvasculature that grows rapidly (from 2 to 7 days) [16], and human tumors (e.g., prostate cancer) that grow over years and are mainly supplied with oxygen and nutriments by pre-existing more mature (i.e., less angiogenic and less permeable) blood vessels [17], co-opted by cancer cells.
In animal models the sprouting angiogenesis is the main mechanism by which tumors acquire a rich microvasculature [1]. The experimental tumors may consist of 40% ECs [18] and the majority of ECs in the neighborhood of a tumor implant are proliferating cells, responding well to the antiangiogenic treatments. By contrast, in many human tumors the microvasculature generally represents only a small fraction of the tumor mass [19] and a minority of ECs are proliferating: 0.15% for prostate cancer [20], 0.7% for clear cell renal cell carcinoma [21], 2.2% for breast cancer [22], 9.9% for colorectal cancer [23], and 10.6% for hepatocellular carcinoma [24]. Moreover, in many cases human tumors can induce their own microvasculature not only by sprouting angiogenesis but also through non-angiogenic mechanisms involving non-proliferating cells (e.g., co-option of pre-existing normal vessels, recruitment of bone-marrow-derived stem cells, and vasculogenic mimicry) that explain the resistance of tumors to conventional anti-angiogenic treatments [25][26][27][28][29][30][31]. These non-angiogenic mechanisms and the resistance mechanisms to common molecularly targeted agents (e.g., angiogenic redundancy, angiogenic dormancy, tumor metabolic adaptation) could explain the frequent inefficacy of the anti-angiogenic treatments with small molecule RTKIs and monoclonal therapeutic antibodies (reviewed in [32][33]).
2. How Anti-Angiogenics Reach Their Target in Cancer: Cellular and Molecular Mechanisms Involved in the Accumulation of Drugs in Tumors
Because the receptors for angiogenic factors are expressed on the abluminal (tissue-facing) plasma membrane of ECs, the small molecule RTKIs and the anti-angiogenic antibodies must cross the endothelial layer of tumor blood vessels. Although small enough to diffuse easily inside tumor tissues (calculated spherical diameter < 1 nm) (https://nanocomposix.eu/pages/molecular-weight-to-size-calculator; accessed on 9 April 2020), the small molecule RTKIs (Table 1) should suffer from a relatively short circulating half-life that limits their therapeutic potential. However, being highly hydrophobic molecules, the RTKIs bind to plasma proteins, especially to albumin (the most abundant plasma protein, which exhibits an average half-life of 19 days), improving their pharmacokinetic profile. Albumin is a protein consisting of 585 amino acids and has a molecular mass of ~ 66 kDa. Because one third of amino acids have an electrical charge (83 positive (lysine and arginine) and 98 negative (glutamic acid and aspartic acid)) albumin has a negative net electric charge (isoelectric point, pI = 5.2) and a high solubility in aqueous solutions at physiological pH [34]. In human plasma, the average plasma concentration is 42 g/L. The presence of 17 covalent disulphide bonds makes human albumin very stable to changes in pH, heat exposure, and denaturing solvents. Based on its inherent biochemical and biophysical properties, the albumin is considered an ideal platform for cancer therapeutic administration [34][35][36].
Table 1. Small molecule anti-angiogenics used in cancer therapy a,b,c.
| RTKI c | Molecular Weight | Albumin Binding | RTK |
|---|---|---|---|
| (Da) | (%) | ||
| Anlotinib | 407.4 | 93.0 | VEGFR, PDGFR, FGFR |
| Apatinib | 397.5 | 92.4 | VEGFR, PDGFR |
| Axitinib | 386.5 | 99.0 | VEGFR, PDGFR |
| Cabozatinib | 501.5 | 99.7 | VEGFR |
| Cediranib | 450.5 | 99.8 | VEGFR, PDGFR |
| Leflunomide | 270.2 | 99.8 | PDGFR |
| Lenvatinib | 523.0 | 99.0 | VEGFR |
| Nilotinib | 529.5 | 93.9 | PDGFR |
| Pazopanib | 437.5 | 99.9 | VEGFR, PDGFR |
| Ponatinib | 532.6 | 99.0 | VEGFR, FGFR |
| Regrorafinib | 482.0 | 99.0 | VEGFR |
| Sorafenib | 464.8 | 99.5 | VEGFR, PDGFR |
| Sunitinib | 398.5 | 95.0 | VEGFR |
| Tivozanib | 454.9 | 99.0 | VEGFR |
| Vandetanib | 475.4 | 90.0 | VEGFR, EGFR |
a PubChem data (https://pubchem.ncbi.nlm.nih.gov/compound; accessed on 9 April 2020). b Upon administration these RTKIs target multiple RTKs including VEGFR, PDGFR, FGFR, and EGFR and may both inhibit angiogenesis and halt tumor cell proliferation. c Please note list is not exhaustive. VEGFR indicates vascular endothelial growth factor receptor; PDGFR, platelet-derived growth factor receptor; FGFR, fibroblast growth factor receptor, EGFR, epithelial growth factor receptor.
Electron microscopy analysis of blood vessels associated with tumor xenograft mouse models for human cancer [37][38][39] has identified caveolae, fenestrae, and vesiculo-vacuolar organelles, three subcellular structures that should be involved in the transendothelial transport of anti-angiogenic molecules (Figure 1A). However, due to the presence of widened (up to 2 µm in diameter) intercellular junctions between tumor endothelial cells [37], and particularly of many large transendothelial channels [40], the actual participation of caveolae, fenestrae, and vesiculo-vacuolar organelles in the transport of anti-angiogenic agents from plasma to the subendothelial space in tumors is very limited. Diffusion mechanisms should therefore allow an extensive and effective transport of small molecule RTKI-albumin conjugates and anti-angiogenic therapeutic antibodies from blood to cancer tissues. The higher the dose of RTKIs and antibodies, the better their efficacy.
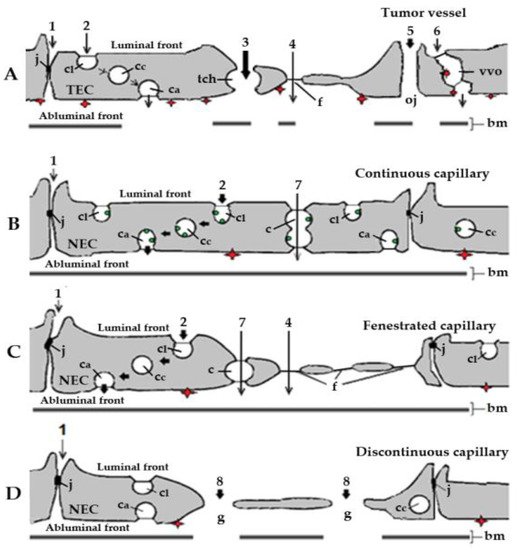
Figure 1. Schematic representation of the basic types of blood capillaries in cancer and healthy organs and tissues. Organelles and routes potentially involved in the transendothelial transport of small molecule receptor tyrosine kinase inhibitors (RTKIs) bound to plasma albumin. (A). Tumor capillary. Effective and efficient transport of RTKI-albumin conjugates by diffusion via large transendothelial channels (tch, pathway 3) and open junctions (pathway 5) towards interstitial space of cancer tissues. By contrast, the actual participation of tight junctions (pathway 1), caveolae (pathway 2), fenestrae (pathway 4), and vesiculo-vacuolar organelles (pathway 6) in the transport of anti-angiogenic agents from plasma to the subendothelial space in tumors should be very limited. (B). Continuous capillary. Caveolar transport (pathway 2)—possibly the main transport mechanism of RTKIs bound to albumin mediated by albumin-binding proteins (ABPs) in continuous capillary endothelia of normal organs and tissues (e.g., skeletal and cardiac muscles, in fat tissue, connective tissue, arterial capillaries of the lung, and in the skin). (C). Fenestrated capillary. Caveolar fluid phase transport (pathway 2; possibly the main transport mechanism of RTKI-albumin conjugates and anti-angiogenic antibodies in fenestrated capillary endothelia of normal organs and tissues). The transport through the diaphragmed fenestrae (pathway 4) should make a minor contribution to the transport of anti-angiogenics due to their negative charges (e.g., fenestrated capillaries in healthy kidney, digestive tract mucosa, and all endocrine glands). (D). Discontinuous capillary. Diffusion through the endothelial gaps (pathway 8; effective and efficient transport but only in liver, spleen, and bone marrow), highly dependent on the concentration of both RTKIs and anti-angiogenic antibodies in blood. The red diamonds represent the angiogenic receptor tyrosine kinases (RTKs) exposed on the abluminal (tissue) front of capillary endothelia. In tumor capillaries, the angiogenic RTKs (the targets of anti-angiogenic agents) are overexpressed. The green dots represent albumin-binding sites (ABPs) associated with caveolae in healthy continuous capillaries. bm, basal membrane; c, channel; ca, abluminal caveolae; cc, cytoplasmic caveolae; cl, luminal caveolae; f, fenestra; g, gap; j, junction; NEC, normal endothelial cell; tch, large transendothelial channel; oj, open junction; TEC, tumor endothelial cell. Adapted with permission from ref. [41]. Copyright 1995 Journal of Endocrinology.
An enhanced permeability of tumor blood vessels, the overexpression of secreted protein acidic and cysteine-rich (SPARC), a matrix-associated protein that binds albumin, and a lack of lymphatic drainage should result in high accumulation of therapeutic antibodies and albumin-RTKI complexes in solid tumors but not into normal tissues and organs [33]. However, factors such as focal necrosis, high interstitial pressure, low microvascular pressure, and the presence of a tumor microvasculature induced by non-angiogenic mechanisms delay extravasation of these anti-angiogenics [42]. This is why an undesirable enhanced transendothelial transport of albumin-RTKI complexes could occur at the level of normal microvasculature, especially in patients with tumors that acquired their microvasculature through non-angiogenic mechanisms. Therefore, the accumulation of albumin-RTKI complexes in healthy organs that may express ABPs could cause serious adverse side effects and toxicity with time [43][44][45].
3. Diffusion and ABP-Mediated Mechanisms Could Underlie the Accumulation of RTKIs Bound to Albumin in Healthy Organs and Tissues
3.1. Endothelial Cell Pathways that May Be Involved in Transport of RTKIs Bound to Albumin
In healthy organs and tissues the exchanges of molecules between the blood and tissues take place at the level of capillaries and postcapillary venules. The main constituent of the wall of capillaries and venules is the endothelium, a single layer of thin flattened ECs that lines their lumens. Morphological studies with the use of tracers visible at the electron microscopic level (reviewed in [46]) have shown that the microvascular ECs are polarized and linked to each other by junctional complexes (tight, adherens, gap) which ensure the continuity and mark the transition between the luminal (blood) and abluminal (tissue) fronts. In contrast to the tumor endothelia, the microvascular endothelium in healthy organs and tissues constitutes a selective barrier for the bidirectional exchanges of macromolecules between the plasma and the interstitial fluid [47][48][49]. Four structures involved in the transendothelial transport of macromolecules in healthy organs and tissues have been defined: intercellular junctions [50][51], caveolae [52], transendothelial channels [53], and endothelial fenestrae [54]. Concerning the presence or absence of fenestrae and discontinuities, three types of capillary endothelia have been classified as continuous (nonfenestrated) (Figure 1B), fenestrated (Figure 1C), and discontinuous (sinusoids) (Figure 1D) (review in [35]). Continuous capillaries are the most common type, and are found in the brain, skeletal and cardiac muscles, in fat tissue, connective tissue, arterial capillaries of the lung, and in the skin.
The endothelial junctions in capillaries of healthy organs and tissues have a low degree of permeability for macromolecules (>2 nm in diameter) [55][56], especially. By contrast, 30% of postcapillary venules and muscular venules exhibit loosely organized endothelial junctions and appear to be permeable to macromolecules having a diameter lower than 5.5 nm [55]. During inflammation, the permeability of the venular junctions is markedly increased and this increased permeability is accompanied by the appearance of numerous gaps (up to 1 µm) between the cells [57] through which plasma molecules including anti-angiogenics drugs (e.g., small molecule RTKIs and therapeutic antibodies) may reach by diffusion healthy tissues and organs.
All ECs contain a population of caveolae (small plasma membrane invaginations 50–70 nm in outer diameter) that move freely from the luminal to the abluminal front of the endothelial cells carrying plasma molecules. The caveolae can take up a bulk of plasma (fluid phase transport) or can carry molecules that have been adsorbed onto the caveolar membrane either electrostatically [58][59][60] or via cognate-specific binding sites [61][62][63][64][65]. The latter mechanism could be involved in the transendothelial export of small molecule RTKIs bound to albumin from blood towards normal tissues in peripheral capillaries with a continuous endothelium (see below).
Transendothelial channels (produced by fusion of several caveolae [66] and/or vesicular invaginations [67]) are open on both the apical and abluminal fronts of the capillary endothelium of normal organs and tissues (Figure 1B,C). They behave as sieves with openings of 20–40 nm. However, the very rare occurrence of such transendothelial channels [66] suggests that their actual participation in the transport of small molecule RTKIs bound to albumin from plasma towards the interstitial space of normal organs and tissues is very limited.
The endothelial fenestrae (Figure 1C), round openings (up to 70 nm in diameter), are found in the fenestrated capillaries of endocrine glands, kidneys, choroid plexus, ciliary bodies, and the mucosa of the gastrointestinal tract [68]. The high density of proteoglycans of high negative charge associated with the luminal aspect of their diaphragms [69][70] should limit the passage through the fenestrae of anionic anti-angiogenics like the small molecule RTKIs bound to albumin.
In the discontinuous capillary endothelia of liver, spleen, and bone marrow, ECs contain large gaps (>100 nm in diameter; Figure 1D) and, in consequence, the endothelium does not impede the transport of anti-angiogenics. Therefore, as in the case of tumor microvessels (see above), diffusion mechanisms allow an extensive and effective accumulation of anti-angiogenics in liver and hematopoietic tissues.
3.2. ABP-Mediated Accumulation of RTKIs Bound to Albumin in Healthy Organs and Tissues Could Be Responsible for Side Effects and Toxicity
The albumin-RTKI complexes form rapidly in the blood and travel through the circulation until they enter the blood vessels of the tumor. Ideally, these complexes should neither interact with, nor be transported through, the microvascular endothelia of healthy organs and tissues. Unfortunately, this is not the case because the drugs that are tightly associated, conjugated, or fused with albumin are kept in circulation and delivered to organs and tissues at a rate and in a similar manner to that of plasma albumin. The capillary endothelium of normal lung, heart, diaphragm, skeletal muscle, and adipose tissue contains three specific albumin-binding glycoproteins (gp18, gp30) [62][63][64], and gp60/albondin [64] restricted mainly to caveolae (Figure 2). Therefore, the accumulation of these drugs bound to albumin could occur not only in tumors but also in normal tissues and organs which, like the tumors, contain SPARC [71]. SPARC is an ABP secreted by various cells whose main function is to mediate interactions between cells and their extracellular matrix during morphogenesis, tissue remodeling, and angiogenesis [72]. Approximately 80% of the total extravascular pool of albumin is equally divided between muscles and the skin [35]. While gp60 is involved in the transendothelial transport of native albumin [64], gp18 and gp30 also act as scavenger receptors that mediate the high affinity endocytosis and degradation of conformationally and chemically modified albumins [73]. Gp18 and gp30 are distributed ubiquitously, being found in the lung, liver, kidney, fat, heart, muscle, brain, adrenal glands, and pancreas [74]. Cultured cells expressing gp30 and gp18 such as the endothelial cells, cardiomyocytes, smooth muscle cells, and fibroblasts are able to bind, internalize, and degrade albumin conformationally modified by endogenous and exogenous ligands including the tyrosine kinase inhibitors. Importantly, chemically modified albumin has a 1000-fold higher affinity for both gp18 and gp30 than native albumin [74], and therefore native albumin should not inhibit the binding, transendothelial transport, accumulation, and degradative process of RTKI-albumin complexes in healthy organs and tissues.
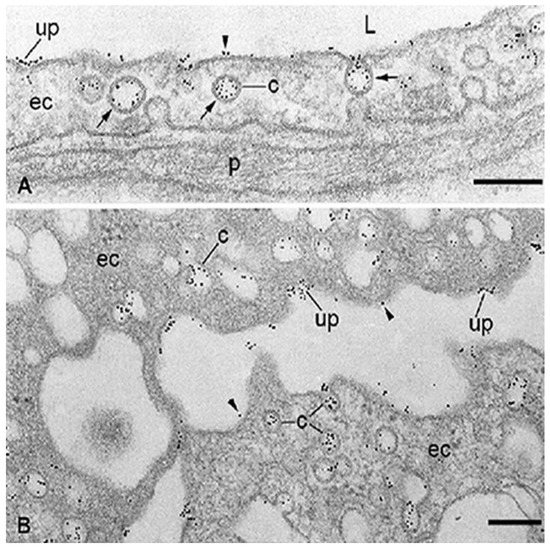
Figure 2. Localization at the electron microscope level of specific binding-sites for albumin associated with the capillary endothelium in healthy adipose tissue (A) and microvascular endothelial cells (ECs) freshly isolated from rat epididymal fat (B). The binding of bovine serum albumin (BSA)-Au5nm conjugate is restricted to the membrane of caveolae (c), predominantly in an adsorptive pattern (arrows) and some uncoated pits (up). Some particles of BSA-Au5nm conjugate rarely appear on plasma membrane proper (arrowhead). ec, endothelial cell; L, capillary lumen; p, pericyte. The scale bar represents 200 nm in both panels. Adapted with permission from ref. [62]. Copyright 1988 Journal of Cell Biology.
Studies on healthy rats have shown that [68Ga]ABY-028, an albumin-binding domain protein-based imaging tracer for positron emission tomography, binds in vivo to albumin, acquires albumin circulatory behavior, and accumulates in liver, spleen, and in muscle tissues [75]. Similar data were published by Park and collaborators [76] who noticed effective accumulation of 64[Cu]-labeled-click chemistry-based albumin nanoplatform conjugates in normal lung, liver, kidney, intestine, and heart with an over four-fold higher uptake than the tumor at 1 h post-injection. Zhong and collaborators [77] showed effective accumulation of anlotinib (an oral RTKI targeting VEGFR, PDGFR, FGFR, and c-Kit; Table 1) in female tumor-bearing mice. The level of tumor exposure to the compound increased as the dose increased. However, after an oral dose of 3 mg/kg of anlotinib the level of tumor exposure was approximately 13.5, 8, and 6.5 times smaller than the level of liver, lung, and kidney, respectively. These data indicate that only the RTKI-albumin complexes that escape the accumulation in normal organs and tissues have the opportunity to interact with the tumors. Because all tissues catabolize albumin [78], the degradation of RTKI-albumin complexes internalized via the caveolar ABPs (i.e., gp18 and gp30) in the cellular lysosomes of healthy organs and tissues may result in a sudden, undesired increase in the free fraction of RTKIs that may accumulate in the cytoplasm of cells and cause adverse effects and toxicity [79][80][81][82][83][84], especially in cases of long-term use of these drugs. The competitive displacement between high-dose administered RTKIs and endogenous compounds such as fatty acids, amino acids, metal ions, hormones, bile acids, and toxic metabolites that share the same binding sites on albumin could also have harmful effects on the body. Moreover, high doses of RTKIs may increase tumor aggressiveness and metastasis [85][86][87]. The accumulation of small molecule RTKIs in normal organs should not be specific for antiangiogenic drugs. Similar ABP-mediated mechanisms are likely to be involved in the accumulation of other therapeutic agents (e.g., chemotherapeutics) bound to albumin in all the healthy tissues and organs where ABPs are expressed. The adverse effects of anti-angiogenesis therapy with small molecule RTKIs include hypertension, diarrhea, fatigue, proteinuria, hand and foot syndrome, thrombocytopenia, and skin and hair discoloration. The combination of small molecule RTKIs bound to albumin with chemotherapeutic agents was thought to improve efficiency and reduce adverse effects on patients. However, this treatment failed to show a survival benefit since the toxicity of the small molecule RTKIs was additive to that of cytotoxic agents [88][89].
In conclusion, the published data suggest that, although albumin improves the pharmacokinetic profile of small molecule inhibitors, it is the binding of drug-albumin complexes to ABPs expressed in healthy tissues and organs of the patients that could be responsible for the side effects and toxicity of RTKIs with time. The development of more specific carriers for small molecule inhibitors than albumin is necessary to enhance efficacy and to reduce the occurrence of adverse effects and toxicities.
This entry is adapted from the peer-reviewed paper 10.3390/diseases9020028
References
- Folkman, J. Tumor angiogenesis factor. Cancer Res. 1974, 34, 2109–2113.
- Bouïs, D.; Kusumanto, Y.; Meijer, C.; Mulder, N.H.; Hospers, G.A. A review on pro- and anti-angiogenic factors as targets of clinical intervention. Pharmacol. Res. 2006, 53, 89–103.
- Raica, M.; Cimpean, A.M. Platelet-derived growth factor (PDGF)/PDGF receptors (PDGFR) axis as target for antitumor and anti-angiogenic therapy. Pharmaceuticals 2010, 3, 572–599.
- Korc, M.; Friesel, R.E. The role of fibroblast growth factors in tumor growth. Curr. Cancer Drug Target 2009, 9, 639–651.
- Presta, M.; Dell’Era, P.; Mitola, S.; Moroni, E.; Ronca, R.; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178.
- Holash, J.; Maisonpierre, P.C.; Compton, D.; Boland, P.; Alexander, C.R.; Zagzag, D.; Yancopoulos, G.D.; Wiegand, S.J. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284, 1994–1998.
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370.
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186.
- Folkman, J. Antiangiogenesis: New concept for therapy of solid tumors. Ann. Surg. 1972, 175, 409–416.
- Boehm, T.; Folkman, J.; Browder, T.; O’Reilly, M.S. Anti-angiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature 1997, 390, 404–407.
- Eberhard, A.; Kahlert, S.; Goede, V.; Hemmerlein, B.; Plate, K.H.; Augustin, H.G. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: Implications for anti-angiogenic tumor therapies. Cancer Res. 2000, 60, 1388–1393.
- Gotink, K.J.; Verheu, H.M.W. Anti-angiogenic tyrosine kinase inhibitors: What is their mechanism of action? Angiogenesis 2010, 13, 1–14.
- van Beijnum, J.R.; Nowak-Sliwinska, P.; Huijbers, E.J.M.; Thijssen, V.L.; Griffioen, A.W. The great escape; the hallmarks of resistance to anti-angiogenic therapy. Pharmacol. Rev. 2015, 67, 441–461.
- Ellis, L.M.; Hicklin, D.J. VEGF-target therapies: Mechanisms of anti-tumor therapy. Nat. Rev. Cancer 2008, 8, 579–591.
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307.
- Zhang, Y.; Toneri, M.; Ma, H.; Yang, Z.; Bouvet, M.; Goto, Y.; Seki, N.; Hoffman, R.M. Real-Time GFP intravital imaging of the differences in cellular and angiogenic behavior of subcutaneous and orthotropic nude-mouse models of human PC-3 prostate cancer. J. Cell Biochem. 2016, 117, 2546–2551.
- Ruoslahti, E. Drug targeting to specific vascular sites. Drug Discov. Today 2002, 7, 1138–1143.
- Algire, G.H.; Chalkley, H.W. Vascular reactions of normal and malignant tissues in vivo. l. Vascular reactions of mice to wounds and to normal and neoplastic transplants. J. Natl. Cancer Inst. 1945, 6, 73–85.
- Dvorak, H.F. Tumor stroma, tumor blood vessels, and antiangiogenesis therapy. Cancer J. 2015, 21, 237–243.
- Vartanian, R.K.; Weidner, N. Endothelial cell proliferation in prostatic carcinoma and prostatic hyperplasia: Correlation with Gleason’s score, microvessel density, and epithelial cell proliferation. Lab. Inv. 1995, 73, 844–850.
- Baldewijns, M.M.; Thijssen, V.L.; Van den Eynden, G.G.; Van Laere, S.J.; Bluekens, A.M.; Roskams, T.; van Poppel, H.; De Bruïne, A.P.; Griffioen, A.W.; Vermeulen, P.B. High-grade clear cell renal cell carcinoma has a higher angiogenic activity than low-grade renal cell carcinoma based on histomorphological quantification and qRT–PCR mRNA expression profile. Br. J. Cancer 2007, 96, 1888–1895.
- Fox, S.B.; Gatter, K.C.; Bicknell, R.; Going, J.J.; Stanton, P.; Cooke, T.G.; Harris, A.L. Relationship of endothelial cell proliferation to tumor vascularity in human breast cancer. Cancer Res. 1993, 53, 4161–4163.
- Vermeulen, P.B.; Verhoeven, D.; Hubens, G.; Van Marck, E.; Goovaerts, G.; Huyghe, M.; De Bruijn, E.A.; Van Oosterom, A.T.; Dirix, L.Y. Microvessel density, endothelial cell proliferation and tumour cell proliferation in human colorectal adenocarcinomas. Ann. Oncol. 1995, 6, 59–64.
- Kim, Y.B.; Park, Y.N.; Park, C. Increased proliferation activities of vascular endothelial cells and tumor cells in residual hepatocellular carcinoma following transcatheter arterial embolization. Histopathology 2001, 38, 160–166.
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603.
- Ribatti, D.; Vacca, A.; Dammacco, F. New non-angiogenesis dependent pathways for tumour growth. Eur. J. Cancer 2003, 39, 1835–1841.
- Kuczynski, E.A.; Reynolds, A.R. Vessel co-option and resistance to anti-angiogenic therapy. Angiogenesis 2020, 13, 55–74.
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493.
- Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Bilecz, A.; Daley, F.; Kostaras, E.; Nathan, M.R.; Wan, E.; Frentzas, S.; Schweiger, T.; et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastases models. J. Pathol. 2017, 241, 362–374.
- Qian, C.-N. Hijacking the vasculature in ccRCC—co-option, remodeling and angiogenesis. Nat. Rev. Urol. 2013, 10, 300–304.
- Frentzas, S.; Simoneau, E.; Bridgeman, V.L.; Vermeulen, P.B.; Fao, S.; Kostaras, E.; Nathan, M.; Wotherspoon, A.; Gao, Z.H.; Shi, Y.; et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat. Med. 2016, 22, 1294–1302.
- Montemagno, C.; Pagès, G. Resistance to anti-angiogenic therapies: A mechanism depending on the time of exposure to the drugs. Front. Cell Dev. Biol. 2020.
- Haibe, Y.; Kreidieh, M.; El Khalief, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance mechanisms to anti-angiogenic therapies in cancer. Front. Oncol. 2020, 10, 221.
- De Sande, L.V.; Cosyns, S.; Willaert, W.; Ceelen, W. Albumin-based cancer therapeutics for intraperitoneal drug delivery: A review. Drug Deliv. 2020, 1, 40–53.
- Sleep, D.; Cameron, J.; Evans, L.R. Albumin as a versatile platform for drug half-life expansions. Biochim. Biophys. Acta 2013, 1830, 5526–5534.
- Yamasaki, K.; Chuang, V.T.; Maruyama, T.; Otagiri, M. Albumin-drug interaction and its clinical implication. Biochim. Biophys. Acta 2013, 1830, 5435–5443.
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between defective endothelial cells explains tumor vessel leakiness to tumor cells. Am. J. Pathol. 2000, 156, 1363–1380.
- Roberts, W.G.; Palade, G.E. Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res. 1997, 57, 765–772.
- Dvorak, A.M.; Kohn, S.; Morgan, E.S.; Fox, P.; Nagy, J.A.; Dvorak, H.F. The vesiculo-vacuolar organelle (VVO): A distinct endothelial cell structure that provides a transcellular pathway for macromolecular extravasation. Leukoc. Biol. 1996, 59, 100–115.
- Sindhwani, S.; Syed, A.M.; Ngai, J.; Kingston, B.R.; Maiorino, L.; Rothschild, J.; Presley Mc, M.; Zhang, Y.; Rajesh, N.U.; Wu, T.; et al. The entry of nanoparticles into solid tumours. Nat. Mater. 2020, 19, 566–575.
- Ghinea, N.; Milgrom, E. Transport of protein hormones through the vascular endothelium. J. Endocrinol. 1995, 145, 1–9.
- Jain, R.K. Transport of molecules across tumor vasculature. Cancer Metastasis Rev. 1987, 6, 559–593.
- Abdel-Qadir, H.; Ethier, J.L.; Lee, D.S.; Thavendiranathan, P.; Amir, E. Cardiovascular toxicity of angiogenesis inhibitors in treatment of malignancy: A systematic review and meta-analysis. Cancer Treat. Rev. 2017, 53, 120–127.
- Dy, G.K.; Adjei, A.A. Understanding, recognizing, and managing of targeted anticancer therapies. Calif. Cancer J. Clin. 2013, 63, 249–279.
- van Heeckeren, W.J.; Ortiz, J.; Cooney, M.M.; Remick, S.C. Hypertension, proteinuria, and antagonism of vascular endothelial growth factor signaling: Clinical toxicity, therapeutic target, or novel biomarker? J. Clin. Oncol. 2007, 25, 2993–2995.
- Simionescu, M.; Ghinea, N. The use of tracers in transport studies. In Models of Lung Disease. Microscopy and Structural Methods; Gill, J., Ed.; Marcel Dekker, Inc.: New York, NY, USA, 1990; pp. 359–408.
- Larson, S.M.; Carrasquillo, J.A.; Reynolds, J.C. Radioimmunodetection and radioimmunotherapy. Cancer Inv. 1984, 2, 358–381.
- Powers, M.R.; Bell, D.R. Initial equilibration of albumin and IgG in rabbit hind paw skin and lymph. Microvasc. Res. 1990, 40, 230–245.
- Ghitescu, L.; Bendayan, M. Transendothelial transport of serum albumin. A quantitative immunocytochemical study. J. Cell Biol. 1992, 117, 745–755.
- Karnovsky, M.J. The ultrastructural basis of capillary permeability studied with peroxidase as a tracer. J. Cell Biol. 1967, 35, 213–236.
- Wissig, S.L.; Williams, M.C. Permeability of muscle capillaries to microperoxidase. J. Cell Biol. 1978, 76, 341–359.
- Bruns, R.R.; Palade, G.E. Studies on blood capillaries. II. Transport of ferritin molecules across the wall of muscle capillaries. J. Cell Biol. 1968, 37, 277–299.
- Simionescu, N.; Simionescu, M.; Palade, G.E. Permeability of muscle capillaries to small heme-peptides. Evidence for the existence of patent transendothelial channels. J. Cell Biol. 1975, 64, 586–607.
- Clementi, F.; Palade, G.E. Intestinal capillaries. I. Permeability to peroxidase and ferritin. J. Cell Biol. 1969, 41, 33–58.
- Simionescu, N.; Simionescu, M.; Palade, G.E. Open junctions in the endothelium of postcapillary venules of the diaphragm. J. Cell Biol. 1978, 79, 27–44.
- Reese, T.S.; Karnovsky, M.J. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J. Cell Biol. 1967, 34, 207–217.
- Majno, G.; Palade, G.E. Studies on inflammation. I. The effect of histamine and serotonin on vascular permeability: An electron microscopic study. J. Biophys. Biochem. Cytol. 1961, 11, 607–626.
- Ghinea, N.; Simionescu, N. Anionized and cationized hemeundecapeptides as probes for cell surface charge and permeability studies: Differentiated labeling of endothelial plasmalemmal vesicles. J. Cell Biol. 1985, 100, 606–612.
- Ghinea, N.; Hasu, M. Charge effect on binding, uptake and transport of ferritin through fenestrated endothelium. J. Submicrosc. Cytol. 1986, 18, 647–659.
- Hervé, F.; Ghinea, N.; Scherrmann, J.M. CNS delivery via adsorptive transcytosis. AAPS J. 2008, 10, 455–472.
- Ghitescu, L.; Fixman, A.; Simionescu, M.; Simionescu, N. Specific binding sites for albumin restricted to plasmalemmal vesicles of continuous capillary endothelium: Receptor-mediated transcytosis. J. Cell Biol. 1986, 102, 1304–1311.
- Ghinea, N.; Fixman, A.; Alexandru, D.; Popov, D.; Hasu, M.; Ghitescu, L.; Eskenasy, M.; Simionescu, M.; Simionescu, N. Identification of albumin-binding proteins in capillary endothelial cells. J. Cell Biol. 1988, 107, 231–239.
- Ghinea, N.; Eskenasy, M.; Simionescu, M.; Simionescu, N. Endothelial albumin binding proteins are membrane-associated components exposed on the cell surface. J. Biol. Chem. 1989, 264, 4755–4758.
- Schnitzer, J.E.; Carley, W.W.; Palade, G.E. Albumin interacts specifically with a 60-kDa microvascular glycoprotein. Proc. Natl. Acad. Sci. USA 1988, 85, 6773–6777.
- Simionescu, M.; Simionescu, N. Endothelial transport of macromolecules: Transcytosis and endocytosis. A look from cell biology. Cell Biol. Rev. 1991, 25, 1–78.
- Frokjaer-Jensen, J. The vesicle controversy. Progress Appl. Microcirc. 1985, 9, 21–42.
- Bundgaard, M.; Frokjaer-Jensen, J.; Crone, C. Endothelial plasmalemmal vesicular profiles as elements in a system of branching invaginations from the cell surface. Proc. Natl. Acad. Sci. USA 1979, 76, 6439–6442.
- Stan, R.V.; Tse, D.; Deharvengt, S.J.; Smits, N.C.; Xu, Y.; Luciano, M.R.; McGarry, C.L.; Buitendijk, M.; Nemani, K.V.; Elgueta, R.; et al. The diaphragms of fenestrated endothelia—gatekeepers of vascular permeability and blood composition. Dev. Cell 2012, 23, 1203–1218.
- Simionescu, M.; Simionescu, N.; Silbert, J.E.; Palade, G.E. Differentiated microdomains on the luminal surface of the capillary endothelium. II. Partial characterization of their anionic sites. J. Cell Biol. 1981, 90, 614–621.
- Pino, R.M. The cell surface of a restrictive fenestrated endothelium. II. Dynamics of cationic ferritin binding and the identification of heparin and heparan sulfate domains on the choriocapillaries. Cell Tissue Res. 1986, 243, 157–164.
- Wang, B.; Chen, K.; Xu, W.; Chen, D.; Tang, W.; Xia, T.S. Integrative genomic analyses of secreted protein acidic and rich in cysteine and its role in cancer prediction. Mol. Med. Rep. 2014, 10, 1461–1468.
- Sage, H.; Vernon, R.B.; Funk, S.E.; Everitt, E.A.; Angello, J. SPARC, a secreted protein associated with cellular proliferation, inhibits cell spreading in vitro and exhibits Ca+2-dependent binding to the extracellular matrix. J. Cell Biol. 1989, 109, 341–356.
- Schnitzer, J.E.; Bravo, J. High affinity binding, endocytosis, and degradation of conformationally modified albumins. Potential role of gp30 and gp18 as novel scavenger receptors. J. Biol. Chem. 1993, 268, 7562–7750.
- Schnitzer, J.; Sung, A.; Horvat, R.; Bravo, J. Preferential interaction of albumin-binding proteins, gp30 and gp18, with conformationally modified albumins. Presence in many cells and tissues with possible role in catabolism. J. Biol. Chem. 1992, 267, 24544–24553.
- Jussing, E.; Lu, L.; Grafström, J.; Tegnebratt, T.; Arnberg, F.; Walllberg Rosik, H.; Wennborg, A.; Holmin, S.; Feldwisch, J.; Stone-Elander, S. [68Ga]ABY-028: An albumin-binding domain (ABD) protein-based imaging tracer for positron emission tomography (PET) studies of altered vascular permeability and predictions of albumin-drug conjugate transport. EJNMMI Res. 2020, 10, 106.
- Park, J.Y.; Song, M.G.; Kim, W.H.; Kim, K.W.; Lodhi, N.A.; Choi, J.Y.; Kim, Y.J.; Kim, J.Y.; Chung, H.; Oh, C.; et al. Versatile and finely tuned albumin nanoplatform based on click chemistry. Theranostics 2019, 9, 3398–3409.
- Zhong, C.C.; Chen, F.; Yang, J.L.; Li, L.; Cheng, C.; Du, F.F.; Zhang, S.P.; Xie, C.Y.; Zhang, N.T.; Olaleye, O.E.; et al. Pharmacokinetics and disposition of anlotinib, an oral tyrosine kinase inhibitor, in experimental animal species. Acta Pharmacol. Sin. 2018, 39, 1048–1063.
- Yedgar, S.; Carew, T.E.; Pittman, R.C.; Beltz, W.F.; Steinberg, D. Tissue sites of catabolism of albumin in rabbits. Am. J. Physiol. 1983, 244, E101–E107.
- Bodnar, R.J. Anti-angiogenic drugs: Involvement in cutaneous side effects and wound-healing complication. Adv. Would Care 2014, 3, 635–646.
- Dobbin, S.J.H.; Cameron, A.C.; Petrie, M.C.; Jones, R.J.; Touyz, R.M.; Lang, N.N. Toxicity of cancer therapy: What the cardiologist needs to know about angiogenesis inhibitors. Heart 2018, 104, 1995–2002.
- Randrup Hansen, C.; Grimm, D.; Bauer, J.; Wehland, M.; Magnusson, N.E. Effects and side effects of using sorafenib and sunitinib in the treatment of metastatic renal cell carcinoma. Int. J. Mol. Sci. 2017, 18, 461.
- Semeniuk-Wojtaś, A.; Lubas, A.; Stec, R.; Szczylik, C.; Niemczyk, S. Influence of tyrosine kinase inhibitors on hypertension and nephrotoxicity in metastatic renal cell cancer patients. Int. J. Mol. Sci. 2016, 17, 2073.
- Sun, Y.; Niu, W.; Du, F.; Du, C.; Li, S.; Wang, J.; Li, L.; Wang, F.; Hao, Y.; Li, C.; et al. Safety, pharmacokinetics, and antitumor properties of anlotinib, an oral multi-target tyrosine kinase inhibitor, in patients with advanced refractory solid tumors. J. Hematol. Oncol. 2016, 9, 105.
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796.
- Ebos, J.M.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 15, 232–239.
- Wang, D.; Xiao, F.; Feng, Z.; Li, M.; Kong, L.; Huang, L.; Wei, Y.; Li, H.; Liu, F.; Zang, H.; et al. Sunitinib facilitates metastatic breast cancer spreading by inducing endothelial cell senescence. Brest Cancer Res. 2020, 22, 103.
- Zhai, W.; Li, S.; Zhang, J.; Chen, Y.; Ma, J.; Kong, W.; Gong, D.; Zheng, J.; Xue, W.; Xu, Y. Sunitinib-suppressed miR-452-5p facilitates renal cancer cell invasion and metastasis through modulating SMAD4/SMAD7 signals. Mol. Cancer 2018, 17, 157.
- Jászai, J.; Schmidt, M.H.H. Trends and challenges in tumor anti-angiogenic therapies. Cells 2019, 8, 1102.
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403.