Protein phosphorylation is a necessary mechanism to drive numerous cellular processes such as cell division, migration, differentiation and programmed cell death. This process is regulated by many enzymes, including cyclin-dependent kinases (CDKs) which phosphorylate proteins on their serine and threonine amino acid residues. The 20 members of CDK family known to this day regulate the cell cycle, transcription and splicing.
- cyclin-dependent kinase inhibitors
- cancer
- cell cycle
- CDKs
- CDK inhibitors
1. Cyclin-Dependent Kinases (CDKs)
Protein phosphorylation is a necessary mechanism to drive numerous cellular processes such as cell division, migration, differentiation and programmed cell death. This process is regulated by many enzymes, including cyclin-dependent kinases (CDKs) which phosphorylate proteins on their serine and threonine amino acid residues. The 20 members of CDK family known to this day regulate the cell cycle, transcription and splicing [1]. A number of kinase inhibitors are emerging every day as potential small molecule drugs, with some of them already being approved by the United States Food and Drug Administration (FDA). Moreover, these already approved kinase targeting drugs now account for more than a quarter of all available drugs [2]. In relation to CDK inhibitors, drugs such as Palbociclib 1, Ribociclib 2 and Abemaciclib 3, have been approved for ER+/HER2- advanced breast cancer treatment [3]. Until recently, the focus of the research was aimed at the highly conserved ATP binding sites of each CDK kinase. Hence, the development of CDK inhibitors has been extremely challenging due to the difficulty of obtaining sufficient selectivity with typical ATP-mimetic compounds. The greatest number of reported compounds has been identified to target the ATP binding pocket. Most recent studies suggest that inhibitors targeting hydrophobic pockets outside the ATP binding site may provide an opportunity for rational target selectivity [4]. Figure 1 illustrates the typical protein structure of the CDK enzyme. The diagram depicts the structural features of a typical kinase domain. Specifically highlighted are the binding pockets of different types of inhibitors, as well as the activation loop.
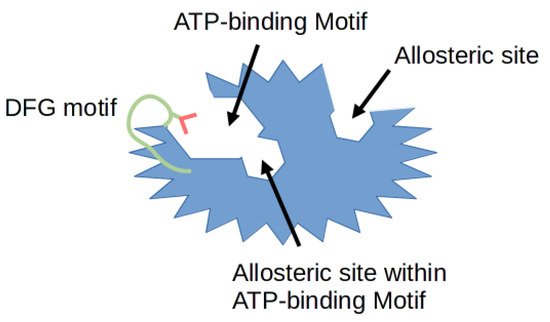
Figure 1. Schematic representation of different types of binding pockets. The protein kinase is shown in blue, with the Asp-Phe-Gly (DFG) motif in green. Red color denotes the aspartate amino acid residue of the DFG motif. The particular regions where different types of inhibitors bind are described below, the allosteric pocket is only a visualization and its place can be anywhere outside the ATP binding site.
2. Cyclin-Dependent Kinase (CDK) Inhibitors in Drug Development
CDK family is known to regulating the cell cycle, transcription and splicing. Deregulation of any of the stages of the cell cycle or transcription leads to apoptosis but, if uncorrected, it can result in a series of diseases such as cancer or neurodegenerative diseases [1,5,6,7].
Within the last 20 years important advances have been achieved in the development of effective strategies to inhibit CDK kinases. Access of the substrate to the active site of CDK kinase is regulated by the activation loop (A-loop) which is very flexible. The A-loop contains between 20–30 amino acids marked by the conserved Asp-Phe-Gly (DFG) tripeptide motif at the proximal end. Phosphorylation of the activation loop activates the kinase. In this state, the DFG sequence fits snugly into a hydrophobic back pocket adjacent to the ATP binding site. Conversely, in the inactive state the DFG motif swings outwards by partially blocking both the ATP and substrate binding pockets [8].
To date, six types of small molecule kinase inhibitors have been defined by the pharmaceutical industry based on their biochemical mechanisms of action (Figure 2). Type I inhibitors interact directly with the ATP binding site and react with the active form of the kinase which is in the DFG-in state and with a phosphorylated activation loop (activation segment). These inhibitors mimic the hydrogen bonds created between the adenine ring of the ATP and the hinge region of the enzyme. Type II inhibitors interact with a DFG-out catalytically inactive conformation of the enzyme and, like type I inhibitors, explore the hinge region and the adenine binding pocket. Type III inhibitors are non-competitive with ATP as they bind to the hydrophobic pocket next to the ATP-binding site, while type IV inhibitors bind away from the ATP binding pocket. Both, type III and IV inhibitors are allosteric in nature [8]. Type V inhibitors interact with two separate regions of the protein kinase domain. This group of inhibitors has been classified as bi-substrate inhibitors. These five classes of inhibitors interact reversibly, while type VI inhibitors form a covalent bond with their target kinase (Figure 2) [9].
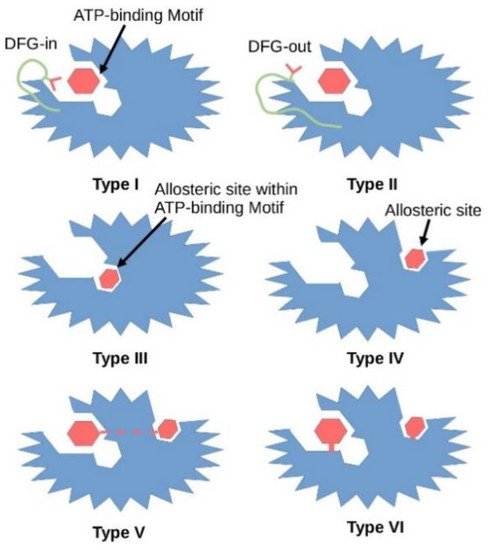
Figure 2. Graphical illustration of different types of kinase inhibitors and their mode of action. Dark red hexagon represents an inhibitor. The protein kinase is shown in blue, the DFG motif in green, the aspartate amino acid residue of the DFG motif in red. In 2015 Wu demonstrated that co-crystal structure of 3-phosphoinositide-dependent protein kinase 1 (PDPK1, PDK1) with ATP showed that type I inhibitors interact with the active conformation of the enzyme where the aspartate residue of the DFG motif points into the ATP binding pocket, while type II inhibitors stabilize the inactive conformation of the enzyme where the aspartate residue faces outward of the binding site (PDB entry: 4RRV). Type III inhibitors interact with the allosteric site within the ATP binding pocket. Type IV inhibitors interact with the allosteric site. However, the allosteric pocket is only a visualization and its place can be anywhere outside the ATP binding site. Type V inhibitors interact with both the allosteric and ATP binding pockets. Type VI inhibitors form covalent bonds with either the ATP binding pocket or the allosteric pocket.
3. Type I Inhibitors
Many heterocyclic compounds can mimic the hydrogen binding motif of adenine, therefore many type I inhibitors have been discovered. As mentioned above, these compounds interact with the ATP-site of the kinase in its active (DFG-in) conformation and with phosphorylated polypeptide region (activation segment) which lies outside the active site pocket. First generation of structurally diverse ATP competitive small molecule type I CDK inhibitors, produced in the late 1990s and early 2000s, have entered clinical trials to treat numerous solid tumors and hematopoietic malignances. Among the list of compounds that have been synthesized as CDK inhibitors, Flavopiridol (Alvocidib) 4 (Figure 3), a flavonoid derived from an indigenous plant from India, is active against CDK1, CDK2, CDK4, CDK6, and CDK9 with IC50 values in the 20–100 nM range (Table 1) [10,11,12,13]. Flavopiridol can inhibit cell cycle progression in G1 as well as G2 phase due to inhibition of CDK2/4 and CDK1 activity, respectively. Early clinical trials proved ineffective because of unsatisfactory efficacy and high toxicity [14,15]. However, later studies confirmed its clinical efficacy in hematological malignancies, and it was granted orphan drug designation for the treatment of patients with acute myeloid leukemia (Table 2) [16].
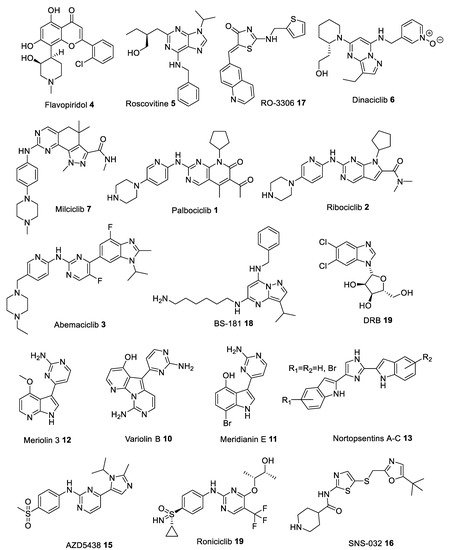
Roscovitine (Seliciclib) 5 (Figure 3), one of the best known CDK inhibitors, is active against CDK2, CDK5, CDK7 and CDK9 (Table 1). This compound is, by far, the most effective inhibitor of CDK5/p25 (IC50 = 160 nM [17]), as shown by numerous studies using this compound as a potential drug against cancer, neurodegenerative or viral diseases, inflammation, polycystic kidney disease (PKD) and glomerulonephritis (Table 2) [18,19,20,21,22]. However, despite many successful preclinical studies, results from several clinical trials are not that promising [23].
Another compound is Dinaciclib 6 (Figure 3), which proved to be a very effective small molecule inhibitor against CDK5 (IC50 = 1 nM [24]) (Table 1). Preclinical studies have shown that Dinaciclib is effective against solid tumors and chronic lymphocytic leukemia (CLL), without adversely affecting T-lymphocytes and their numbers (Table 2) [25].
Moreover, Milciclib 7 (Figure 3), an orally bioavailable inhibitor of cyclin-dependent kinases (CDKs) and several other protein kinases responsible for controlling cell growth and replication, has recently obtained the orphan drug designation for thymic carcinoma. It is currently under investigation as a potential drug target for treatment of glioma and hepatocellular carcinoma (HCC) (Table 2) [26,27]. It inhibits CDK2 with IC50 of 45 nM and exhibits submicromolar activity against other CDKs including CDK1, CDK4 and CDK5 resulting in a block in the G1 (gap) phase of the cell cycle (Table 1) [28]. Furthermore, Milciclib was found to reduce levels of microRNAs, miR-221 and miR-222, which promote the formation of blood supply (angiogenesis) in cancer tumors [29].
And finally, Palbociclib 1 and Ribociclib 2 (Figure 3), novel CDK4/6 inhibitors, were approved as effective drugs against HR+/HER2- metastatic breast cancer (Table 2) [30,31]. They selectively inhibit CDK4/6 (Table 1), thereby inhibiting retinoblastoma (Rb) protein phosphorylation early in the G1 phase leading to cell cycle arrest, causing defects in DNA replication and efficiently suppress cancer cell proliferation. Most recent data show that both drugs demonstrate a synergistic effect when combined with other drugs, for example Palbociclib and aromatase inhibitor Letrozole [32], Ribociclib and either anaplastic lymphoma kinase (ALK) inhibitor or the mitogen-activated protein kinase kinase (MAP2K, MEK) inhibitor Trametinib [33]. Moreover, utilizing this approach leads to a significant reduction in the development of resistance during prolonged treatment courses [31].
In addition, Tamoxifen 8 has been found to be effective against breast cancer. It reduces CDK5 activity by interacting with p25 and p35, thus preventing CDK5 activation. Tamoxifen can also lower Tau protein phosphorylation, which may suggest that tamoxifen could be used against Alzheimer’s disease [34].
Yet another inhibitor, 5,6-dichlorobenzimidazone-1-β-D-ribofuranoside (DRB) 9 (Figure 3) possesses high selectivity against CDK9, with nearly 25-fold difference in potency over CDK2 and CDK7 (Table 1) [35]. In HeLa cells, DRB (75 μM) inhibited 60-75% of nuclear heterogeneous RNA (hnRNA) synthesis. DRB inhibited a HeLa protein kinase which phosphorylated an RNA polymerase II-derived peptide [36]. DRB can also inhibit HIV transcription (IC50 = ~4 μM) by targeting elongation enhanced by the HIV-encoded transactivator Tat (Table 2) [37].
Table 1. Kinase inhibitory activities of type I CDK inhibitors.
| Inhibitor | Kinase IC50 [nM] | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| CDK1/B | CDK2/A | CDK2/E | CDK4/D | CDK5/p25 | CDK6/D | CDK7/H | CDK8/C | CDK9/T1 | |
| Flavopiridol 4 [38,39] | 30 | 100 | 100 | 20–40 | - | 60 | 110 | - | 20 |
| Roscovitine 5 [40] | 650 | 700 | 700 | >100,000 | 160 | >100,000 | 460 | >100,000 | 600 |
| RO-3306 17 [41] | 35 | - | 340 | >2000 | - | - | - | - | - |
| Dinaciclib 6 [42] | 3 | 1 | 1 | 100 | 1 | - | - | - | 4 |
| Milciclib 7 [28] | 398 | 45 | 363 | 160 | 265 | - | 150 | - | - |
| Palbociclib 1 [43] | >10,000 | >10,000 | >10,000 | 11 | >10,000 | 15 | - | - | - |
| Ribociclib 2 [44] | 113,000 | 76,000 | 76,000 | 10 | 43,900 | 39 | - | - | - |
| Abemaciclib 3 [45] | 1627 | - | 504 | 2 | 355 | 10 | 3910 | - | 57 |
| BS-181 18 [46] | 8100 | 730 | 880 | 33,000 | 3000 | 47,000 | 21 | - | 4200 |
| DRB 9 [47] | 17,000 | - | >10,000 | >10,000 | - | - | >10,000 | >10,000 | 340 |
| Meriolin 3 12 [48] | 170 | 11 | - | >100,000 | 170 | >100,000 | >100,000 | - | 6 |
| Variolin B 10 [49] | 60 | 80 | - | >10,000 | 90 | >10,000 | >1000 | - | 26 |
| Meridianin E 11 [50] | 180 | 800 | 1800 | 3000 | 150 | - | - | - | 18 |
| Nortopsentins 13 [51] | 310–900 | - | - | - | - | - | - | - | - |
| AZD5438 15 [52] | 16 | 45 | 6 | 449 | 14 | 21 | 821 | - | 20 |
| Roniciclib 19 [53] | 7 | - | 9 | 11 | - | - | 25 | - | 5 |
| SNS-032 16 [54] | 480 | 38 | 48 | 925 | 340 (CDK5/p35) |
- | 62 | - | 4 |
Table 2. Type I CDK inhibitors at different phases of clinical and pre-clinical studies. Trial information obtained from ClinicalTrials.gov as of January 2021.
| Inhibitor | Main Targets | Condition or Disease | Phase | Status | Identifier |
|---|---|---|---|---|---|
| Flavopiridol 4 | CDK1, CDK2, CDK4, CDK6, CDK9 | Acute Myeloid Leukemia (AML) | on the market | “orphan drug” | - |
| Roscovitine 5 | CDK2, CDK7, CDK9 | Pituitary Cushing Disease | II | active | NCT02160730 NCT03774446 |
| Cystic Fibrosis | II | terminated | NCT02649751 | ||
| Advanced Solid Tumors | I | terminated | NCT00999401 | ||
| Lung Cancer | II | terminated | NCT00372073 | ||
| RO-3306 17 [41] | CDK1 | Acute Myeloid Leukemia (AML) | pre-clinical | - | - |
| Dinaciclib 6 | CDK1, CDK2, CDK5, CDK9 | Chronic Lymphocytic Leukemia (CLL) | on the market | “orphan drug” | - |
| Breast and Lung Cancers | II | terminated | NCT00732810 | ||
| Milciclib 7 | CDK1, CDK2, CDK4, CDK7 | Hepatocellular Carcinoma (HCC) | II | active | NCT03109886 |
| Thymic Carcinoma | II | terminated | NCT01301391 NCT01011439 |
||
| Palbociclib 1 | CDK4, CDK6 | HR+/HER2- Breast Cancer | on the market | used in combination with Letrozole | - |
| III | active, to be used with other drugs like Fulvestrant | NCT02692755 | |||
| Head and Neck, Brain, Colon, and other Solid Cancers | II | active, to be used alone and in combination with different drugs | NCT02255461 NCT03446157 NCT02896335 NCT03965845 |
||
| Ribociclib 2 | CDK4, CDK6 | HR+/HER2- Breast Cancer | on the market | used in combination with Letrozole | - |
| III | active, to be used with other drugs like Fulvestrant | NCT02422615 NCT03439046 NCT03294694 |
|||
| Prostate, and other Solid Cancers | II | active, to be used alone and in combination with different drugs | NCT02555189 NCT01543698 NCT02934568 |
||
| Abemaciclib 3 | CDK4, CDK6 | HR+/HER2- Breast Cancer | on the market | used in combination with Fulvestrant | - |
| III | active, to be used with other drugs like Letrozole | NCT02763566 | |||
| Lung, Brain, Colon, and other Solid Cancers | II or III | active, to be used alone and in combination with different drugs | NCT04545710 NCT02152631 NCT03220646 NCT04616183 NCT03310879 |
||
| BS-181 18 [46] | CDK7 | Breast, Lung, Prostate and Colorectal Cancers | pre-clinical | - | - |
| DRB 9 [55] | CDK7, CDK8, CDK9 | HIV Transcription | pre-clinical | - | - |
| Meriolin 3 12 [48] | CDK1, CDK2, CDK5, CDK9 | Neuroblastoma, Glioma, Myeloma, Colon Cancer | pre-clinical | - | - |
| Variolin B 10 [56] | CDK1, CDK2, CDK5, CDK9 | Murine Leukemia | pre-clinical | - | - |
| Meridianin E 11 [57] | CDK1, CDK5, CDK9 | Larynx Carcinoma, Myeloid Leukemia | pre-clinical | - | - |
| Nortopsentins 13 [58] | CDK1 | Malignant Pleural Mesothelioma (MPM) | pre-clinical | - | - |
| AZD5438 15 | CDK1, CDK2, CDK5, CDK6, CDK9 | Advanced Solid Malignancies | I | terminated | NCT00088790 |
| Roniciclib 19 | CDK1, CDK2, CDK4, CDK7, CDK9 | Lung and Advanced Solid Cancers | II | terminated | NCT02161419 NCT01573338 NCT02656849 |
| SNS-032 16 | CDK2, CDK7, CDK9 | Chronic Lymphocytic Leukemia and other Solid Cancers | I | terminated | NCT00446342 NCT00292864 |
Novel alkaloids, acting as CDK inhibitors, were also found in some marine organisms. Variolins, 7-azaindole based alkaloids isolated from the antarctic sponge Kirkpatrickia variolosa [59,60], showed in vitro activity against a murine (P388) leukemia cell line with submicromolar potencies by preventing cell proliferation, and inducing apoptosis (Table 2) [56,59]. Variolin B 10 (Figure 3), in particular, was found to inhibit CDK1 and CDK2 kinases, in the micromolar concentration range (Table 1) [61]. Meridianins A-G, a family of 3-(2-aminopyrimidine)indoles, which originate from the ascidian Aplidium meridianum, were demonstrated to inhibit several protein kinases, especially Meridianin E 11 (Figure 3), which can selectively inhibit CDK1 and CDK5 in the low micromolar range (Table 1) [62]. Based on the latter two compounds, Meriolins 12, a new class of inhibitors, have been designed. These new derivatives have been reported to strongly inhibit various protein kinases, especially CDK1, CDK2, CDK4 and CDK9 (Table 1) [48]. Most recent analysis provides a high potential of Meriolins in the treatment of cancer and noncancer pathologies such as polycystic kidney disease, neurodegenerative diseases, stroke, chronic inflammation, and bipolar disorders (Table 2) [48]. Nortopsentins A-C 13 (Figure 3), antifungal 1,4-bisindolylimidazole marine alkaloids, having an imidazole as a spacer between the two indole units, isolated from the Caribbean deep sea Spongosorites ruetzleri, displayed in vitro cytotoxicity against P388 leukemia cells (IC50 4.5–20.7 µM). Analogues in which the imidazole ring of the alkaloid was replaced by other five or six membered heterocycles were able to inhibit the activity of the cyclin-dependent kinase 1 (CDK1) with submicromolar IC50 values (in particular 3-[(2-indolyl)-5-phenyl]-pyridines, phenyl-thiazolyl-7-azaindoles, indolyl-thiazolyl-4-azaindole and indolyl-thiazolyl-7-azaindole derivatives) (Table 1) [51]. Preliminary results indicate, that Nortopsentins, and their analogues, were active against malignant pleural mesothelioma (MPM), a very aggressive human malignancy poorly responsive to currently available therapies (Table 2) [58].
Recent development has enabled combinatorial treatment regimens which can demonstrate synergistic anticancer mechanisms. For instance, THZ1 14 (Figure 7) a covalent CDK7 inhibitor, was found to selectively downregulate CDK7-mediated phosphorylation of RNA polymerase II, indicative of transcriptional inhibition. Further investigations revealed that the survival of triple negative breast cancer (TNBC) cells relied heavily on the B-cell lymphoma 2 (BCL-2)/B-cell lymphoma-extra large (BCL-XL) signaling axes in cells. Thus, combining the CDK7 inhibitor THZ1 with the BCL-2/BCL-XL inhibitors (ABT-263/ABT199) offer a preclinical proof to significantly improve the poor prognosis in TNBC [63].
However, the complexity of CDK biology and the undesired toxicity related to the off-target effects of the existing pan-CDK inhibitors, led to decisions by several pharmaceutical companies to discontinue the development of many potential anti-cancer agents, exampled with AZD5438 15, Roniciclib, SNS-032 16, RO-3306 17, BS-181 18 and Roniciclib 19 (Figure 3) (Table 1 and Table 2) [64,65,66]. Therefore, new classes of more selective CDK inhibitors, with strong potential to deliver a meaningful therapeutic impact, were needed.
One of those compounds is CDK5 inhibitory protein (CIP), a small protein which contribute to nerve cells’ degeneration. CIP specifically blocks the hyperactivated state of CDK5 only when it is linked to p25/p29, while allowing normal activation of CDK5 by p35/p39. The selective inhibition of p25/CDK5 hyperactivation in vivo, through overexpression of CIP, reduced neurodegeneration and improved cognitive function of transgenic mice, without affecting normal neurodevelopment [67]. These findings suggest that CIP could possibly be used to selectively inhibit the p25/CDK5 hyperactivation as a potential therapeutic target to treat certain cancers caused by aberrant CDK5 activation.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22062806