Metal oxide nanoparticles (NPs) have received a great deal of attention as potential theranostic agents. Despite extensive work on a wide variety of metal oxide NPs, few chemically active metal oxide NPs have received Food and Drug Administration (FDA) clearance. The clinical translation of metal oxide NP activity, which often looks so promising in preclinical studies, has not progressed as rapidly as one might expect. The lack of FDA approval for metal oxide NPs appears to be a consequence of the complex transformation of NP chemistry as any given NP passes through multiple extra- and intracellular environments and interacts with a variety of proteins and transport processes that may degrade or transform the chemical properties of the metal oxide NP. Moreover, the translational models frequently used to study these materials do not represent the final therapeutic environment well, and studies in reduced preparations have, all too frequently, predicted fundamentally different physico-chemical properties from the biological activity observed in intact organisms. Understanding the evolving pharmacology of metal oxide NPs as they interact with biological systems is critical to establish translational test systems that effectively predict future theranostic activity.
- cell trafficking
- endocytosis
1. Introduction
There is tremendous interest in metal oxide nanoparticles (NPs) for use in therapeutic applications such as diagnostic tools and drugs in which the nanoparticles are either the active agent or passive, drug delivery nanocarriers. To-date, there are over thirty different metal oxide formulations being studied that may have biological effects [1], but few have garnered FDA clearance. While nanomaterials have demonstrated potential therapeutic benefit in many biomedical applications, clinical translation of individual formulation has not progressed as rapidly as one would expect given the plethora of preclinical studies [2][3]. We believe that the slow progression to approved drugs may result, in part, from the types of translational models used to study these materials, and the emerging evidence that the activity of nanomaterials in cell-free conditions and reduced preparations can be fundamentally different from the biological properties of the nanomaterial when studied in either cell culture conditions or, more importantly, in intact organisms. A better understanding of the evolving pharmacology of metal oxide nanoparticles (NPs) as they interact with biological systems is critical to establish translational test systems that can effectively predict future drug potential.
2. The Origin of Biological Activity in the Structure of Nanoparticles
All nanoparticles, regardless of elemental composition or shape, have extremely high surface area:volume ratios that confer chemical reactivity not observed in particles with larger dimensions (i.e., > 100 nm) [4]. The solubility of nanomaterials in biological fluids is dictated by surface composition, surface charge, and the hydrophobicity/hydrophilicity profile. The surface electrostatic interactions between particles determine their propensity to aggregate and adsorb proteins to their surface. The chemical and biological reactivity as well as biodistribution of the nanomaterials are derived from these fundamental properties.
2.1. Redox Reactivity of Metal Oxides
Metal oxides NPs have an ability to participate in myriad biologically important redox reactions and mimic a wide range of enzymes including catalases, oxidases, dismutases, peroxidases, ATPases and phosphatases [5][6][7][8]. The native enzymes expressing similar redox activity play manifold and crucial roles in redox-dependent signaling cascades, and metal oxide NPs can disrupt or restore redox balance in cells through these reactions and signaling processes [8].Not all metal oxides exhibit the same enzymatic activities, and mimetic activities can be ‘biased’ by the local environment surrounding the particle. There are three key factors that determine the interaction between the biological milieu and the redox activity at the nano-bio interface: the half-cell potential of the elements comprising the particle, the organization of the surface atoms of the nanoparticle, and the oxidation state of the ions on and within the nanoparticle. Reduction in the oxidation number of the metal (i.e., the accounting of the number of electrons a metal possess or lacks) occurs when the crystal loses an oxygen atom and forms a vacancy in the NP. Thermodynamically, any oxide is potentially reducible [9], and the distinction between reducible and non-reducible metal oxides depends on the ease with which oxygen vacancies can be formed [9]. In non-reducible metal oxides, the thermodynamic cost of formation of oxygen vacancies is high, and redox activity is absent [10]. In reducible metal oxides, oxygen vacancy formation is thermodynamically more favorable and occurs at lattice surfaces and edges where the coordination number of the surface atoms (i.e., the total number of bonds to the atom) is less than inside the crystalline structure of the oxide. The edge is also where lattice strain is highest [11]; all of which facilitates the formation of oxygen vacancies [9][12]. Thus, the highest enzyme-mimetic activity occurs at the surface of the nanoparticle [9][13]. Many transition-metal oxides, such as TiO2, MnOx, NiO, Fe2O3, and CeO2, are reducible because the energetic barriers to oxygen vacancy formation are low, and vacancies can occur spontaneously across the surface of the crystal.The chemical mechanisms underlying redox activity can be divided into either electrophilic or nucleophilic reactions. Extra-facial, adsorbed oxygen is responsible for most of the electrophilic reactions, whereas interfacial oxygen, where lattice oxygen vacancies are created, underlie the nucleophilic reactions [14]. In general, nucleophilic oxygen (e.g., the oxide ion) is capable of carrying out selective oxidations while it seems that electrophilic oxygen species, which are deficient in electrons (e.g., the superoxide radical), appear to be more promiscuous and are largely responsible for non-selective oxidation [15]. There is usually a ‘preferred’ or stable oxidation state in each NP, and surface defects created by spontaneous loss of oxygen result in different valence states (i.e., Ce4+ > Ce3+). The redox state of the metal oxide can flip-flop repeatedly between valance states, which provide durable, recycling, catalytic activity.The mechanisms of cyclic, regenerative redox reactions have been studied in cerium oxide NPs because of the relatively low barrier for the transition between Ce4+  Ce3+. Cerium oxide demonstrates both superoxide dismutase and catalase mimetic activity [16][17][18]. In the reaction scheme shown below, the hydroxyl radical is the ‘seed’ for the balanced set of redox processes. Given the high oxidation potential of •OH (+2.31 V at pH 7.0) and a high rate constant, hydroxyl radical can react rapidly with many biomolecules and lead to oxidative damage. However, the superoxide anion is likely the reactant with ceria. Superoxide anion is continually produced as a result of aerobic respiration, and the production of this reactive oxygen species, which functions as an important biological signaling molecule, can be greatly up-regulated in disease states. Superoxide dismutase converts superoxide anion into peroxide, and this species is quickly converted to water and oxygen by catalase or by reacting with hydroxyl anions via Fenton reactions. Both of these potent oxidizing agents, O2− and H2O2, likely contribute to oxidative stress and damage of DNA, proteins, and lipids [19].Formation of oxygen vacancies within the ceria nanoparticle lattice structure is central to this regenerative mimetic activity. The sequence of proposed reactions to explain the mimetic activity of cerium oxide is shown below [20][21]:
Ce3+. Cerium oxide demonstrates both superoxide dismutase and catalase mimetic activity [16][17][18]. In the reaction scheme shown below, the hydroxyl radical is the ‘seed’ for the balanced set of redox processes. Given the high oxidation potential of •OH (+2.31 V at pH 7.0) and a high rate constant, hydroxyl radical can react rapidly with many biomolecules and lead to oxidative damage. However, the superoxide anion is likely the reactant with ceria. Superoxide anion is continually produced as a result of aerobic respiration, and the production of this reactive oxygen species, which functions as an important biological signaling molecule, can be greatly up-regulated in disease states. Superoxide dismutase converts superoxide anion into peroxide, and this species is quickly converted to water and oxygen by catalase or by reacting with hydroxyl anions via Fenton reactions. Both of these potent oxidizing agents, O2− and H2O2, likely contribute to oxidative stress and damage of DNA, proteins, and lipids [19].Formation of oxygen vacancies within the ceria nanoparticle lattice structure is central to this regenerative mimetic activity. The sequence of proposed reactions to explain the mimetic activity of cerium oxide is shown below [20][21]: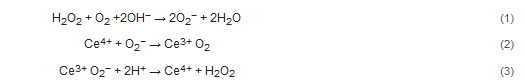

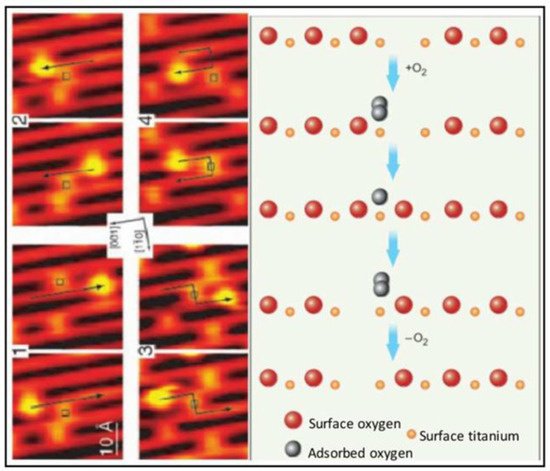 Figure 1. Scanning tunneling microscopic (STM) images (left) and molecular schematics (right) demonstrate the interactions of molecular oxygen adsorbed to the surface of titanium oxide at the site of oxygen vacancies within the crystal structure of the metal oxide. Each adjacent STM image shows the surface structure of the metal oxide before and after the interaction with molecular oxygen and the migration of the oxygen vacancy. Used with permission from Pinto et al. [9].In addition to reducing concentrations of oxidizing agents, most metal oxides can elicit free radical-mediated toxicity via the formation of hydroxyl through Fenton-type reactions [22][23]. Within reactive sites generated at oxygen vacancies, electron donor or acceptor regions interact with molecular O2 to form O2•—which in turn can generate additional reactive oxygen species (ROS). Fe3O4 magnetic nanoparticles, for example, exhibited intrinsic peroxidase-like activity under acidic conditions and a catalase-like activity at neutral pH [24]. Moreover, both hematite nano-Fe2O3 and maghemitenano-Fe2O3 induced hydroxyl radical formation in more acidic environments through Fenton reactions. The specific reaction that predominates (i.e., oxidation or reduction) will depend on the valence state of the crystal, which is modified by the pH of the cellular compartment in which the particle resides (as shown above for cerium oxide). Redox cell damage may also occur if dissociation of the metal ions (i.e., Ag NPs and Quantum Dots) elicits cellular enzyme deactivation, membrane disruption, altered electron transfer, reduced mitochondrial membrane potentials, or changes in gene expression; all of which may increase the accumulation of cellular oxidants [25][26][27][28][29][30][31].The potential benefits of metal oxide nanoparticles for medical applications have emerged from their robust antioxidant properties [32][33][34]. Most studies fail to parse the impact of the local environment on nanoparticle reactivity and concentrate on the net effect of the nanoparticle as either pro- or anti-oxidant. This creates the (mistaken) impression that the metal oxide exhibits only one type of redox reactivity when in reality metal oxide NPs may have flexible redox reactivity that can be biased toward oxidation or reduction depending on the valence state and the milieu of the nanoparticle (pH, protein corona, cell-free media, serum, cell culture media, etc.).
Figure 1. Scanning tunneling microscopic (STM) images (left) and molecular schematics (right) demonstrate the interactions of molecular oxygen adsorbed to the surface of titanium oxide at the site of oxygen vacancies within the crystal structure of the metal oxide. Each adjacent STM image shows the surface structure of the metal oxide before and after the interaction with molecular oxygen and the migration of the oxygen vacancy. Used with permission from Pinto et al. [9].In addition to reducing concentrations of oxidizing agents, most metal oxides can elicit free radical-mediated toxicity via the formation of hydroxyl through Fenton-type reactions [22][23]. Within reactive sites generated at oxygen vacancies, electron donor or acceptor regions interact with molecular O2 to form O2•—which in turn can generate additional reactive oxygen species (ROS). Fe3O4 magnetic nanoparticles, for example, exhibited intrinsic peroxidase-like activity under acidic conditions and a catalase-like activity at neutral pH [24]. Moreover, both hematite nano-Fe2O3 and maghemitenano-Fe2O3 induced hydroxyl radical formation in more acidic environments through Fenton reactions. The specific reaction that predominates (i.e., oxidation or reduction) will depend on the valence state of the crystal, which is modified by the pH of the cellular compartment in which the particle resides (as shown above for cerium oxide). Redox cell damage may also occur if dissociation of the metal ions (i.e., Ag NPs and Quantum Dots) elicits cellular enzyme deactivation, membrane disruption, altered electron transfer, reduced mitochondrial membrane potentials, or changes in gene expression; all of which may increase the accumulation of cellular oxidants [25][26][27][28][29][30][31].The potential benefits of metal oxide nanoparticles for medical applications have emerged from their robust antioxidant properties [32][33][34]. Most studies fail to parse the impact of the local environment on nanoparticle reactivity and concentrate on the net effect of the nanoparticle as either pro- or anti-oxidant. This creates the (mistaken) impression that the metal oxide exhibits only one type of redox reactivity when in reality metal oxide NPs may have flexible redox reactivity that can be biased toward oxidation or reduction depending on the valence state and the milieu of the nanoparticle (pH, protein corona, cell-free media, serum, cell culture media, etc.).
2.2. Intracellular pH Environments and Metal Oxide NP Activity
Fan et al. [32] synthesized Pt-Ft nanoparticles using an apoferritin protein shell/scaffold as a nanoreactor to control the synthesis of size-tunable Pt nanostructures. One to two nm Pt–Ft NPs synthesized in this way possessed both catalase and peroxidase activities. However, these superparamagnetic iron particles (SPIONs) demonstrated peroxidase activity in acidic solutions, but lost this activity in more neutral solutions and instead expressed catalase-like activity through a series of coupled reactions [32]. The antioxidant properties of CeOx NPs dominate at physiological pH, whereas these particles exhibit high oxidase activity at acidic pH [35], likely related to a net shift in the valence of material to Ce4+ [36]. Moreover, SOD activity is enhanced at lower pH relative to catalase activity, resulting in the accumulation of peroxide [37]. In more neutral conditions, CeOx NPs display both SOD and catalase activity [16]. Silver NPs were similarly sensitive to pH: the level of hydroxyl radical formation through a Fenton-like mechanism was dependent on pH-hydroxyl radical formation occurred at pH 4.6 or lower, but at more neutral pH, no significant formation of hydroxyl radicals occurred [38]. Thus, the tuning or biasing of the enzymatic mimesis of metal oxide NPs is modulated by intracellular pH, which can vary both by cellular localization (i.e., cytosol versus lysosome; see Figure 2) or whether the cells are immortalized or not [32][39].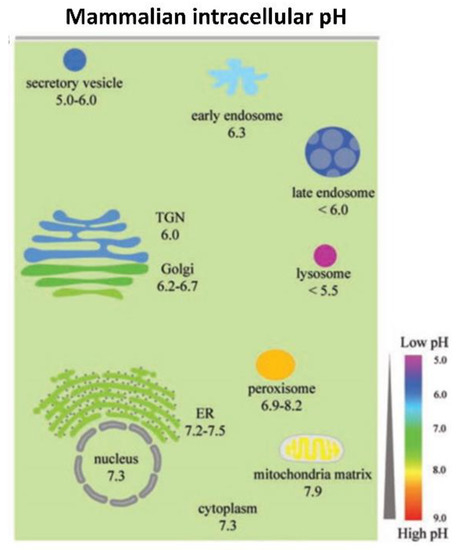 Figure 2. The range of pH values in intracellular compartments is shown schematically. The extent and type of chemical activity of metal oxide NPs may vary significantly, even in a single cell, across a wide range of pH values in different organelles. ER, endoplasmic reticulum and TGN, trans-Golgi network. From Shen et al. [40] with permission.
Figure 2. The range of pH values in intracellular compartments is shown schematically. The extent and type of chemical activity of metal oxide NPs may vary significantly, even in a single cell, across a wide range of pH values in different organelles. ER, endoplasmic reticulum and TGN, trans-Golgi network. From Shen et al. [40] with permission.
2.3. Model System Effects
The biological effects of nanoparticles depend not just on the properties of the material in standardized conditions, but also on the biological system in which the nanoparticles are active [41][42][43][44][45][46]. There is increasing evidence that immortalized cells (i.e., differentiated cancer cells) have unique redox profiles that are different from their native, healthy counterparts [47][48]. Selective cytoprotection has been reported following administration of nanoceria in normal, healthy cells, but not in cancer cells [49][50]. Often, cancer cells rely more on glycolysis for energy production, and consequently they maintain more acidic intracellular pH values [51]. Where additional protons are present (i.e., lactate accumulation or localization in acidic organelles), Ce3+ reacts with a H+ and O2•− to produce Ce4+ and H2O2, leading to net oxidation [37][52]. Moreover, in a comparison of immortalized colorectal cells (HCT 116) and human embryonic kidney (HEK 293) cells, CeOx NPs increased the ROS load and subsequently induced apoptosis in colon cancer cells but not in the embryonic kidney cells, suggesting that differences in either cellular localization or baseline pH existed in these cell types [53]. The accumulation of CeOx NPs in this study was not evaluated, so it is possible that the amount of material taken up by these two cell types could have differed and impacted ROS formation. In a study of three different MnOx NPs (MnO2, Mn3O2, Mn3O4) with different valance states, the biological implications of valence switching were examined in a cell-free system. Each MnOx NP exhibited both pro- and anti-oxidant activities simultaneously, including oxidase-, catalase-, and superoxide dismutase (SOD)-like activities. These MnOx NPs decreased cell viability in a dose-dependent manner in colorectal adenocarcinoma cells (Caco-2) regardless of valence, and the largest reduction in viability was associated with Mn3O4 > Mn3O2 > MnO2. While the MnOx NPs were all cytotoxic, they protected cells when the cells were challenged with peroxide—suggesting that catalase mimetic activity was protective [54]. Unlike many other metal oxides, the MnOx NPs were devoid of peroxidase or hydroxyl radical scavenging activity in cell-free assays, but when studied in cells, the MnOx NPs were located in the cytosol, which has a higher pH than most other organelles in the cell, and the local pH may have biased the enzyme mimetic activities of the different valences and allowed the particles to provide cytoprotective activity when the cells were challenged with peroxide. Consistent with these findings, MnOx nanoparticles increased catalase and SOD activities, while they also decreased glutathione levels in cell culture [55]. The decreases in cell viability caused by MnOx NPs were associated with mitochondrial dysfunction and apoptosis, presumably secondary to the reduction in glutathione levels. Glutathione is critical to maintain mitochondrial function and cell viability, and loss of sufficient glutathione levels in mitochondria increased oxidative stress [56]. Most often, MnOx NPs are cytotoxic in immortalized cell cultures, but the outcome of administration of these materials in whole animals is variable, and some studies show that they are safe (Xiao et al., 2013) but not others [57]. Hence, these nanoparticles may be protective in certain redox states and certain cell types but not others.The variable redox effects of metal oxide NPs, which may be either pro-or antioxidant, have been vexing. Beyond the effects of the cells studied and the impact of pH in these test systems, redox activity of NPs may be related to the manner of synthesis (valence ratio), the size of the particles, the complement of adsorbed proteins, and the cellular localization of the material. The redox activity of metal oxide NPs is not easily predicted since local environments may vary so much. Moreover, findings in cell-free systems are not fully recapitulated in more representative biological environments like cell culture or intact animals. The biological impact of these materials seems to be tied to the baseline redox status of the cells being studied, which adds yet another source of variability when trying to characterize the likely therapeutic effect of nanoparticles. While many disease states elevate oxidative stress in tissues, not all tissues will have the same redox changes driven by the disease state. Thus, even within a single organism, the redox activity of a nanoparticle may differ organ by organ or even organelle by organelle. Since the delivery of metal oxides occurs passively, these materials distribute widely throughout the body including healthy cells, and healthy cells may be negatively impacted by NPs while the benefit of these materials as antioxidants may be observed only in cells that have a high oxidative load [47][48]. Understanding how these factors modify redox reactivity will be critical to the future development of therapeutic nanoparticles [58][59].
This entry is adapted from the peer-reviewed paper 10.3390/antiox10040547
References
- Mirshafiee, V.; Sun, B.; Chang, C.H.; Liao, Y.P.; Jiang, W.; Jiang, J.; Liu, X.; Wang, X.; Xia, T.; Nel, A.E. Toxicological Profiling of Metal Oxide Nanoparticles in Liver Context Reveals Pyroptosis in Kupffer Cells and Macrophages versus Apoptosis in Hepatocytes. ACS Nano 2018, 12, 3836–3852.
- Hua, S.; de Matos, M.B.C.; Metselaar, J.M.; Storm, G. Current Trends and Challenges in the Clinical Translation of Nanoparticulate Nanomedicines: Pathways for Translational Development and Commercialization. Front. Pharm. 2018, 9, 790.
- Luxenhofer, R.; Barz, M.; Schillmeier, M. Quo vadis nanomedicine? Nanomedicine 2014, 9, 2083–2086.
- Oberdorster, G. Safety assessment for nanotechnology and nanomedicine: Concepts of nanotoxicology. J. Intern. Med. 2010, 267, 89–105.
- Sims, C.M.; Hanna, S.K.; Heller, D.A.; Horoszko, C.P.; Johnson, M.E.; Montoro Bustos, A.R.; Reipa, V.; Riley, K.R.; Nelson, B.C. Redox-active nanomaterials for nanomedicine applications. Nanoscale 2017, 9, 15226–15251.
- Chen, W.; Li, S.; Wang, J.; Sun, K.; Si, Y. Metal and metal-oxide nanozymes: Bioenzymatic characteristics, catalytic mechanism, and eco-environmental applications. Nanoscale 2019, 11, 15783–15793.
- Grimaud, A.; Diaz-Morales, O.; Han, B.; Hong, W.T.; Lee, Y.L.; Giordano, L.; Stoerzinger, K.A.; Koper, M.T.M.; Shao-Horn, Y. Addendum: Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 2017, 9, 828.
- Singh, S. Nanomaterials Exhibiting Enzyme-Like Properties (Nanozymes): Current Advances and Future Perspectives. Front. Chem. 2019, 7, 46.
- Pinto, F.M.; Suzuki, V.Y.; Silva, R.C.; La Porta, F.A. Oxygen Defects and Surface Chemistry of Reducible Oxides. Front. Mater. 2019, 6, 260.
- Puigdollers, A.R.; Schlexer, P.; Tosoni, S.; Pacchioni, G. Increasing Oxide Reducibility: The Role of Metal/Oxide Interfaces in the Formation of Oxygen Vacancies. ACS Catal. 2017, 7, 6493–6513.
- Feng, B.; Sugiyama, I.; Hojo, H.; Ohta, H.; Shibata, N.; Ikuhara, Y. Atomic structures and oxygen dynamics of CeO2 grain boundaries. Sci. Rep. 2016, 6, 20288.
- Esch, F.; Fabris, S.; Zhou, L.; Montini, T.; Africh, C.; Fornasiero, P.; Comelli, G.; Rosei, R. Electron localization determines defect formation on ceria substrates. Science 2005, 309, 752–755.
- Nolan, M.; Fearon, J.E.; Watson, G.W. Oxygen vacancy formation and migration in ceria. Solid State Ion. 2006, 177, 3069–3074.
- Fernández--García, M.; Rodriguez, J.A. Metal Oxide Nanoparticles. In Encyclopedia of Inorganic and Bioinorganic Chemistry; Scott, R.A., Ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2011.
- Reddy, B.M. Redox properties of metal oxides. In Metal Oxides: Chemistry and Applications; Fierro, J.L.G., Ed.; CRC Press: Boca Raton, FL, USA, 2005; pp. 215–246.
- Heckert, E.G.; Karakoti, A.S.; Seal, S.; Self, W.T. The role of cerium oxide redox state in the SOD mimetic activity of nanoceria. Biomaterials 2008, 29, 2705–2709.
- Pirmohamed, T.; Dowding, J.M.; Singh, S.; Wasserman, B.; Heckert, E.; Karakoti, A.S.; King, J.E.S.; Seal, S.; Self, W.T. Nanoceria exhibit redox state-dependent catalase mimetic activity. ChemComm 2010, 46, 2736–2738.
- Walkey, C.D.; Das, S.; Seal, S.; Erlichman, J.S.; Heckman, K.; Ghibelli, L.; Traversa, E.; McGinnis, J.F.; Self, W.T. Catalytic properties and biomedical applications of cerium oxide nanoparticles. Environ. Sci. Nano 2015, 15.
- Thomas, C.; Mackey, M.M.; Diaz, A.A.; Cox, D.P. Hydroxyl radical is produced via the Fenton reaction in submitochondrial particles under oxidative stress: Implications for diseases associated with iron accumulation. Redox. Rep. 2009, 14, 102–108.
- Celardo, I.; Traversa, E.; Ghibelli, L. Cerium oxide nanoparticles: A promise for applications in therapy. J. Exp. Oncol. 2011, 9, 47–51.
- Reed, K.; Bush, N.; Burns, Z.; Doherty, G.; Foley, T.; Milone, M.; Maki, K.L.; Cromer, M. Modeling the Kinetic Behavior of Reactive Oxygen Species with Cerium Dioxide Nanoparticles. Biomolecules 2019, 9, 447.
- Fubini, B.; Hubbard, A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic. Biol. Med. 2003, 34, 1507–1516.
- Huang, Y.W.; Wu, C.H.; Aronstam, R.S. Toxicity of Transition Metal Oxide Nanoparticles: Recent Insights from in vitro Studies. Materials 2010, 3, 4842–4859.
- Wang, B.; Yin, J.J.; Zhou, X.Y.; Kurash, I.; Chai, Z.F.; Zhao, Y.L.; Feng, W.Y. Physicochemical Origin for Free Radical Generation of Iron Oxide Nanoparticles in Biomicroenvironment: Catalytic Activities Mediated by Surface Chemical States. J. Phys. Chem. C 2013, 117, 383–392.
- Carlson, C.; Hussain, S.M.; Schrand, A.M.; Braydich-Stolle, L.K.; Hess, K.L.; Jones, R.L.; Schlager, J.J. Unique cellular interaction of silver nanoparticles: Size-dependent generation of reactive oxygen species. J. Phys. Chem. B 2008, 112, 13608–13619.
- Holt, K.B.; Bard, A.J. Interaction of silver(I) ions with the respiratory chain of Escherichia coli: An electrochemical and scanning electrochemical microscopy study of the antimicrobial mechanism of micromolar Ag+. Biochemistry 2005, 44, 13214–13223.
- Jiang, W.; Kim, B.Y.; Rutka, J.T.; Chan, W.C. Nanoparticle-mediated cellular response is size-dependent. Nat. Nanotechnol. 2008, 3, 145–150.
- Kang, S.J.; Lee, Y.J.; Lee, E.K.; Kwak, M.K. Silver nanoparticles-mediated G2/M cycle arrest of renal epithelial cells is associated with NRF2-GSH signaling. Toxicol. Lett. 2012, 211, 334–341.
- Liu, T.; Xiao, B.; Xiang, F.; Tan, J.; Chen, Z.; Zhang, X.; Wu, C.; Mao, Z.; Luo, G.; Chen, X.; et al. Ultrasmall copper-based nanoparticles for reactive oxygen species scavenging and alleviation of inflammation related diseases. Nat. Commun. 2020, 11, 2788.
- Nguyen, K.C.; Rippstein, P.; Tayabali, A.F.; Willmore, W.G. Mitochondrial Toxicity of Cadmium Telluride Quantum Dot Nanoparticles in Mammalian Hepatocytes. Toxicol. Sci. Off. J. Soc. Toxicol. 2015, 146, 31–42.
- Zhang, Y.; Gu, A.Z.; Xie, S.; Li, X.; Cen, T.; Li, D.; Chen, J. Nano-metal oxides induce antimicrobial resistance via radical-mediated mutagenesis. Environ. Int. 2018, 121, 1162–1171.
- Fan, J.; Yin, J.J.; Ning, B.; Wu, X.; Hu, Y.; Ferrari, M.; Anderson, G.J.; Wei, J.; Zhao, Y.; Nie, G. Direct evidence for catalase and peroxidase activities of ferritin-platinum nanoparticles. Biomaterials 2011, 32, 1611–1618.
- Heckman, K.; DeCouteau, W.; Estevez, A.Y.; Reed, K.; Constanzo, W.; Sanford, D.; Leiter, J.C.; Clauss, J.; Knapp, K.; Gomez, C.; et al. Custom Cerium Oxide Nanoparticles Protect against a Free Radical Mediated Autoimmune Degenerative Disease in the Brain. ACS Nano 2013, 7, 10582–10596.
- Valgimigli, L.; Baschieri, A.; Amorati, R. Antioxidant activity of nanomaterials. J. Mater. Chem. B 2018, 6, 2036–2051.
- Kim, H.Y.; Ahn, J.K.; Kim, M.I.; Park, K.S.; Park, H.G. Rapid and label-free, electrochemical DNA detection utilizing the oxidase-mimicking activity of cerium oxide nanoparticles. Electrochem. Commun. 2019, 99, 5–10.
- Asati, A.; Santra, S.; Kaittanis, C.; Nath, S.; Perez, J.M. Oxidase-like activity of polymer-coated cerium oxide nanoparticles. Angew. Chem. Int. Ed. Engl. 2009, 48, 2308–2312.
- De Marzi, L.; Monaco, A.; De Lapuente, J.; Ramos, D.; Borras, M.; Di Gioacchino, M.; Santucci, S.; Poma, A. Cytotoxicity and genotoxicity of ceria nanoparticles on different cell lines in vitro. Int. J. Mol. Sci. 2013, 14, 3065–3077.
- He, W.; Zhou, Y.T.; Wamer, W.G.; Boudreau, M.D.; Yin, J.J. Mechanisms of the pH dependent generation of hydroxyl radicals and oxygen induced by Ag nanoparticles. Biomaterials 2012, 33, 7547–7555.
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61.
- Shen, J.; Zeng, Y.; Zhuang, X.; Sun, L.; Yao, X.; Pimpl, P.; Jiang, L. Organelle pH in the Arabidopsis endomembrane system. Mol. Plant 2013, 6, 1419–1437.
- Al Faraj, A.; Shaik, A.P.; Shaik, A.S. Effect of surface coating on the biocompatibility and in vivo MRI detection of iron oxide nanoparticles after intrapulmonary administration. Nanotoxicology 2015, 9, 825–834.
- Hauser, A.K.; Mitov, M.I.; Daley, E.F.; McGarry, R.C.; Anderson, K.W.; Hilt, J.Z. Targeted iron oxide nanoparticles for the enhancement of radiation therapy. Biomaterials 2016, 105, 127–135.
- Lunov, O.; Syrovets, T.; Buchele, B.; Jiang, X.E.; Rocker, C.; Tron, K.; Nienhaus, G.U.; Walther, P.; Mailander, V.; Landfester, K.; et al. The effect of carboxydextran-coated superparamagnetic iron oxide nanoparticles on c-Jun N-terminal kinase-mediated apoptosis in human macrophages. Biomaterials 2010, 31, 5063–5071.
- Mahmoudi, M.; Laurent, S.; Shokrgozar, M.A.; Hosseinkhani, M. Toxicity Evaluations of Superparamagnetic Iron Oxide Nanoparticles: Cell “Vision” versus Physicochemical Properties of Nanoparticles. ACS Nano 2011, 5, 7263–7276.
- Muthiah, M.; Park, I.K.; Cho, C.S. Surface modification of iron oxide nanoparticles by biocompatible polymers for tissue imaging and targeting. Biotechnol. Adv. 2013, 31, 1224–1236.
- Sabareeswaran, A.; Ansar, E.B.; Harikrishna Varma, P.R.; Mohanan, P.V.; Kumary, T.V. Effect of surface-modified superparamagnetic iron oxide nanoparticles (SPIONS) on mast cell infiltration: An acute in vivo study. Nanomedicine 2016, 12, 1523–1533.
- Jorgenson, T.C.; Zhong, W.; Oberley, T.D. Redox imbalance and biochemical changes in cancer. Cancer Res. 2013, 73, 6118–6123.
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253.
- Corsi, F.; Caputo, F.; Traversa, E.; Ghibelli, L. Not Only Redox: The Multifaceted Activity of Cerium Oxide Nanoparticles in Cancer Prevention and Therapy. Front. Oncol. 2018, 8, 309.
- Gao, Y.; Chen, K.; Ma, J.L.; Gao, F. Cerium oxide nanoparticles in cancer. Oncotargets Ther. 2014, 7, 835–840.
- Shamim, U.; Hanif, S.; Albanyan, A.; Beck, F.W.J.; Bao, B.; Wang, Z.W.; Banerjee, S.; Sarkar, F.H.; Mohammad, R.M.; Hadi, S.M.; et al. Resveratrol-induced apoptosis is enhanced in low pH environments associated with cancer. J. Cell. Physiol. 2012, 227, 1493–1500.
- Xu, C.; Qu, X.G. Cerium oxide nanoparticle: A remarkably versatile rare earth nanomaterial for biological applications. NPG Asia Mater. 2014, 6.
- Datta, A.; Mishra, S.; Manna, K.; Das Saha, K.; Mukherjee, S.; Roy, S. Pro-Oxidant Therapeutic Activities of Cerium Oxide Nanoparticles in Colorectal Carcinoma Cells. ACS Omega 2020, 5, 9714–9723.
- Jiang, X.; Gray, P.; Patel, M.; Zheng, J.; Yin, J.J. Crossover between anti- and pro-oxidant activities of different manganese oxide nanoparticles and their biological implications. J. Mater. Chem. B 2020, 8, 1191–1201.
- Alarifi, S.; Ali, D.; Alkahtani, S. Oxidative Stress-Induced DNA Damage by Manganese Dioxide Nanoparticles in Human Neuronal Cells. Biomed. Res. Int. 2017, 2017, 5478790.
- Mari, M.; Morales, A.; Colell, A.; Garcia-Ruiz, C.; Kaplowitz, N.; Fernandez-Checa, J.C. Mitochondrial glutathione: Features, regulation and role in disease. Biochim. Biophys. Acta 2013, 1830, 3317–3328.
- Singh, S.P.; Kumari, M.; Kumari, S.I.; Rahman, M.F.; Kamal, S.S.; Mahboob, M.; Grover, P. Genotoxicity of nano- and micron-sized manganese oxide in rats after acute oral treatment. Mutat. Res. 2013, 754, 39–50.
- Corbo, C.; Molinaro, R.; Parodi, A.; Toledano Furman, N.E.; Salvatore, F.; Tasciotti, E. The impact of nanoparticle protein corona on cytotoxicity, immunotoxicity and target drug delivery. Nanomedicine 2016, 11, 81–100.
- Nakayama, T.; Tanaka, T.; Shiraki, K.; Hase, M.; Hirano, A. Suppression of single-wall carbon nanotube redox reaction by adsorbed proteins. Appl. Phys. Express 2018, 11, 075101.